Маккан-Олбрайт синдром (MAS)
Синдром Мак-Кьюна—Олбрайта—Брайцева (СМОБ) относится к редким формам церебрального преждевременного полового развития (ППР) в сочетании с фиброзной дисплазией костей и асимметричной пигментацией кожи; описан в 1907 г. как болезнь displasia fibrosa polycistia (фиброзная остеодистрофия).
Синдром характеризуется триадой симптомов: фиброзно-кистозной дисплазией костной ткани; асимметричной пигментацией кожных покровов; разнообразными эндокринопатиями, наиболее частой из которых является гонадотропнозависимое преждевременное половое созревание (Петеркова В.А. и др., 1999).
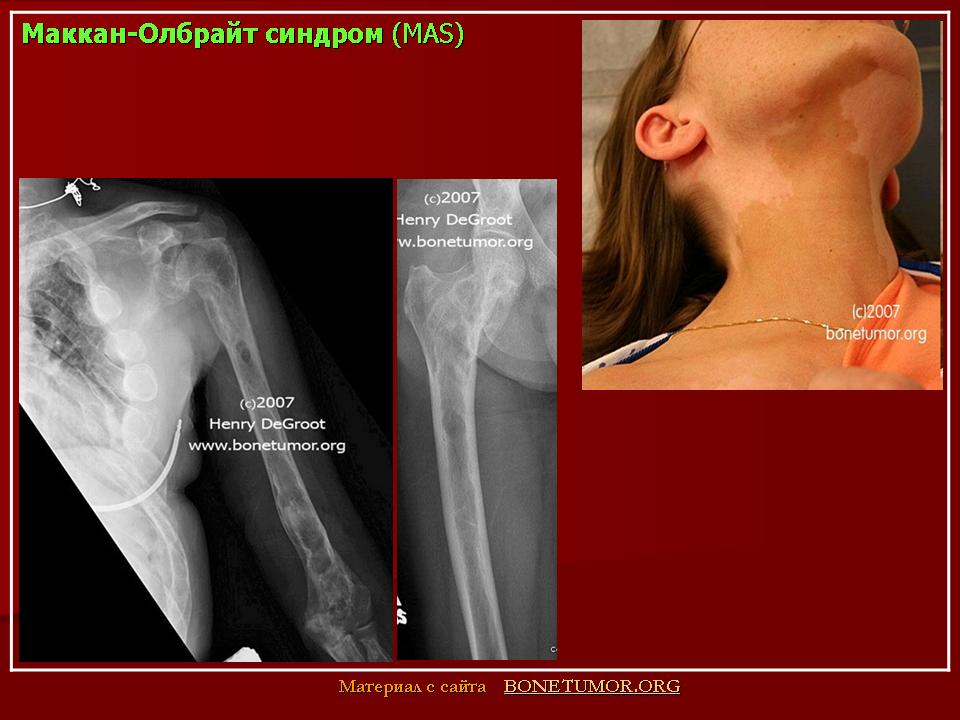
Причину возникновения данного синдрома авторы связывают с мутацией в гене, кодирующем а-субъединицу гуанидинтрифосфат-связывающего белка, принадлежащего к классу стимулирующих (Gsa). Мутация возникает спорадически на постзиготной стадии. Маркером синдрома является пигментация кожи светло-кофейного цвета, неправильной формы, обычно на груди, спине, в области поясницы и бедер.
Эти пятна присутствуют с рождения или распространяются по мере поста ребенка. При обследовании 9 пациенток с клинической картиной СМОБ на молекулярно-генетическом уровне выявлено наличие характерной мутации в участке ДНК, кодирующем молекулу аргинина в 201 положении. Кроме того, при тяжелой и среднетяжелой клинике СМОБ авторам удалось обнаружить мутацию в периферических лейкоцитах — при отсутствии при этом синдроме симптомов со стороны красной крови и кроветворных органов. Авторы предполагают, согласно теории о постзиготном характере мутации при СМОБ, что чем раньше в периоде эмбриогенеза произошла мутация, тем шире распределяется популяция клеток, содержащих мутантную ДНК.
Заболевание встречается только у девочек и представляет врожденную наследственную патологию. Во всей мировой литературе не описано ни одного случая заболевания мальчиков (Сметник В.П., Тумилович Л.Г., 1997). Наиболее тяжелые проявления при этом заболевании — костные нарушения.
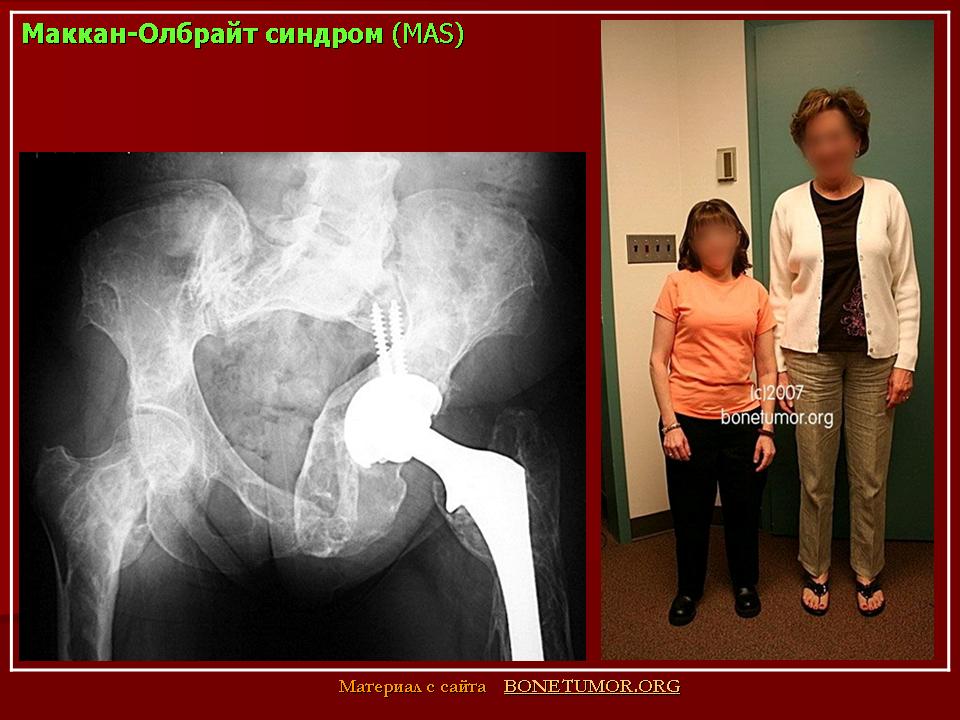
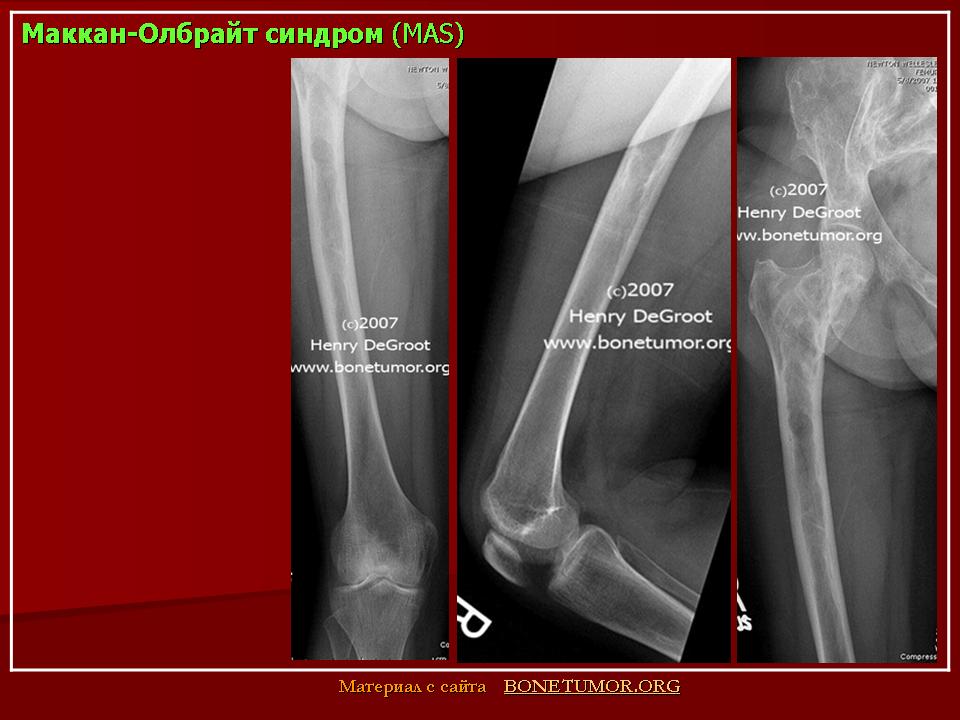
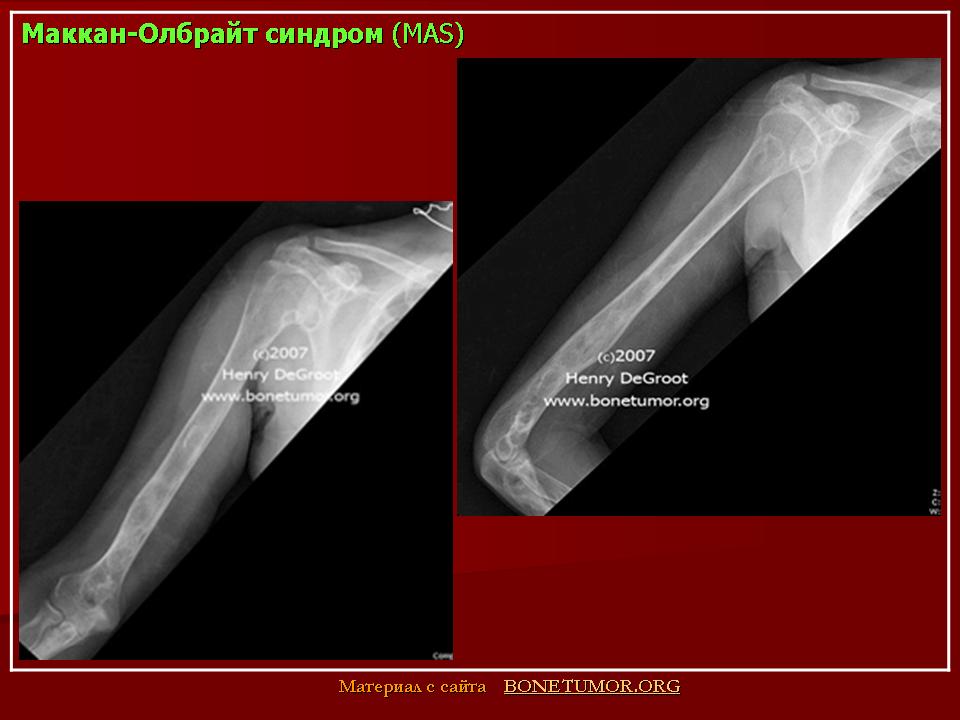
Причины развития костной патологии труднообъяснимы, поскольку она развивается на фоне повышения уровня эстрогенов, усиливающих процесс кальцинации костей и костеобразования (Сметник В.П., Тумилович Л.Г., 1997). По мнению S.J. Magalini et al. (1981), фиброзно-кистозная дисплазия — фокальное замещение кортикального слоя пролиферирующими фибробластами, чаще поражаются длинные трубчатые кости. Наличие кист приводит к деформации кости, возникновению патологических переломов с искривлением нижележащего края конечности.
Наиболее типичной является деформация бедра, имеющего вид «пастушьего посоха». Берцовая кость чаще искривляется по вальгус-ному типу. Кроме того, формирование фиброзных кист возможно в костях лицевого скелета и основания черепа. При этом возникают лицевая асимметрия, односторонний экзофтальм, в редких случаях наблюдаются неврологические и психические нарушения (атрофия зрительного нерва, потеря слуха, судороги, задержка психического развития). Процесс патологии костной ткани и частота переломов снижаются после завершения пубертата (Брайцев В.Р., 1954).
Клиника эндокринной патологии проявляется в автономной гиперфункции гипофиза и периферических желез внутренней секреции (Петеркова В.А. и др., 1999).
Среди эндокринных нарушений наиболее частым является преждевременное половое развитие. Последнее начинается в возрасте от 6—9 мес. жизни до 7-летнего возраста и может проявиться в двух формах — неполной и полной. Для полной формы ППР характерно развитие двух половых признаков — менархе и увеличение молочных желез, для неполной — наличие вторичных половых признаков при отсутствии менструаций. Характерным является ускорение роста в длину. По достижении репродуктивного возраста женщины с этой патологией по росту и телосложению не отличаются от женщин, у которых половое развитие началось своевременно. Если при полной форме ППР темп полового развития ускорен, то при неполной он удлинен и иногда превосходит время физиологического развития в 2 раза (Мороз М.Г., 1987).
В отличие от истинных форм ППР при синдроме Мак-Кью-на—Олбрайта—Брайцева не отмечается полового оволосения, диф-ференцировка скелета ускоряется незначительно. Яичники обычно не увеличены и содержат крупные, длительно персистирующие фолликулярные кисты. Уровень гонадотропных гормонов обычно не превышает нормы. Выраженность этой патологии снижается после завершения пубертата. В механизм его вовлечена рецептор-нозависимая аденилат-циклиновая система неспецифической активации, которая стимулирует гормонообразование без воздействия тропных гормонов.
Уровень гонадотропных гормонов в крови, по данным большинства исследователей, не превышает возрастной нормы. Реакция гонадотропных гормонов на введение люлиберина также соответствует допубертатным значениям (Kaufmann M.A. et al., 1986).
Однако имеются сообщения и о повышенной секреции ЛГ и ФСГ при данном заболевании, особенно при длительно существующих симптомах преждевременного полового развития. В подобных случаях предполагается вторичное созревание гипоталамо-гипофа-зарной системы в результате длительной гиперэстрогенной сенсибилизации гонад отрофов.
Имеются сообщения о ряде других эндокринных нарушений, сопровождающих синдром СМОБ, среди которых следует отметить акромегалию, гиперпаратиреоз, гипертиреоз, синдром Иценко—Кушинга и витамин-D-резистентный рахит (Danon M., 1975; Micaki M., 1988; Richard L., 1989).
Следовательно, данная патология по этиологическим факторам и по патогенезу остается не полностью изученной.
Больные данной группы нуждаются в комплексном обследовании и консультации гинеколога-эндокринолога, лучевого диагноста, эндокринолога, офтальмолога, травматолога, а также в определении уровня тропных и гонадотропных гормонов.


Маккан-Олбрайт синдром (MAS)