Врожденное заболевание, описанное R. Pfeiffer в 1964 году, которое характеризуется акроцефалической формой черепа, синдактилией пальцев кистей и стоп и гипертрофией больших пальцев кистей. Синдром подразделяется на три типа.
• Тип I характеризуется классическим внешним видом, проявляющимся краниосиностозом, широкими большими пальцами кисти и синдактилией, а также нормальным умственным развитием. Данная форма совместима с жизнью.
• Тип II - череп в виде «трилистника», проптоз глазных яблок, широкие большие пальцы кисти, различные аномалии внутренних органов, анкилоз локтевого сустава и тяжелые мальформации центральной нервной системы. Данная форма обычно сопровождается ранней гибелью.
• Тип III - краниосиностоз, выраженный проптоз глазных яблок, отсутствие формы черепа в виде «трилистника», анкилоз локтевого сустава, различные аномалии внутренних органов. У пораженных плодов отмечаются тяжелые неврологические нарушения с неблагоприятным прогнозом и ранней гибелью.
Распространенность. Неизвестна. Со времени опубликования первого описания имеются сообщения приблизительно о 30 клинических наблюдениях.
Этиология. Тип I имеет аутосомно-доминантное наследование или возникает вследствие новых мутаций. Типы II и III встречаются спорадически.
Риск рецидива. Различный, зависит от этиологии. В тех случаях, когда заболевания наследуются аутосомно-доминантно, то риск составляет 50%. Если же синдром возникает вследствие новых мутаций, то риск рецидива очень низок.
Диагностика. Эхографические признаки синдрома Пфайффера (Pfeiffer) включают черепно-лицевые аномалии (брахицефалия, акроцефалия, краниосиностоз за счет преждевременного закрытия коронарного шва, гипертелоризм, маленький нос и глубокая переносица), аномалии кистей и стоп (частичная синдактилия II и III пальцев кисти; II, III и IV пальцев стопы; широкие большие пальцы кистей и гипертрофия больших пальцев стопы).
Генетические нарушения. Синдром Пфайффера (Pfeiffer) генетически гетерогенен. Некоторые случаи связаны с мутациями гена FGFR1, локализованного в хромосоме 8р11.22-р12. Также имеются сообщения о мутациях гена FGFR2, который располагается в хромосоме 10q25-q26.
Сочетанные аномалии. Атрезия хоан, трахео- и бронхомаляция, череп в виде «трилистника», слияние позвонков, мальформация Арнольда-Киари (Arnold-Chiari), гидроцефалия, неперфорированный анус. После рождения могут возникать судороги и задержка умственного развития.
Дифференциальный диагноз. Основной дифференциальный диагноз проводится с синдромами, которые характеризуются краниосиностозами (синдром Апера (Apert), Карпентера (Carpenter), Крузона (Crouzon), аномалия Kleeblattschadel и танатофорная дисплазия).
Прогноз. Прогноз зависит от тяжести сочетанных аномалий, особенно от степени выраженности неврологическоих поражений. При типе I в большинстве случаев прогноз благоприятный, в то время как типы II и III практически несовместимы с жизнью и приводят к ранней гибели.
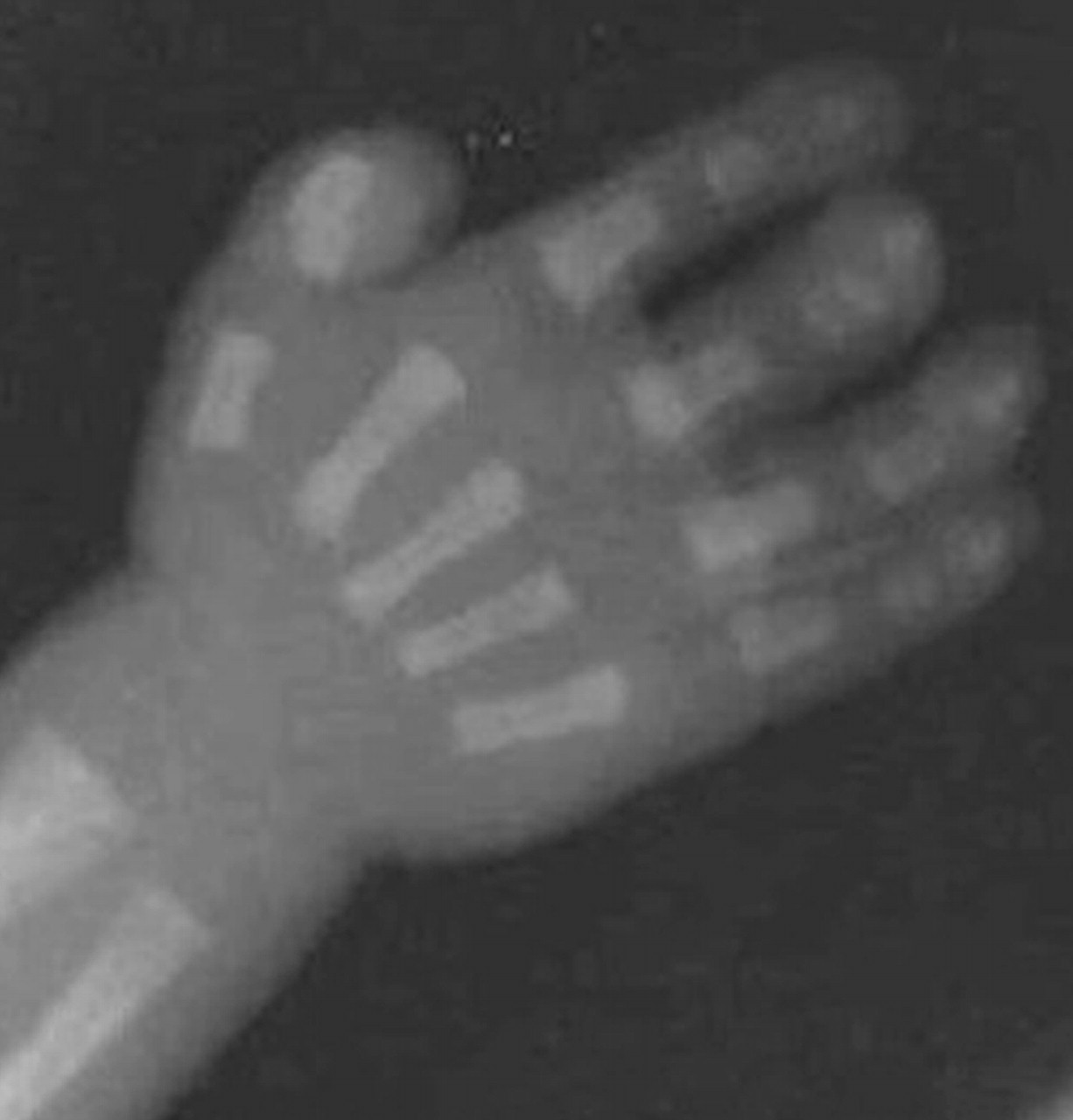
Синдром Пфайффера встречается примерно в 1 на 100000 новорожденных. Возникает данный синдром в результате спонтанной мутации в гене FGRFIили II. Ряд авторов считают, что синдром может возникнуть у детей от пожилых отцов. Характерен аутосомно-доминантный тип наследования этого заболевания. То есть достаточно, чтобы один из родителей имел повреждённый ген, чтобы передать его потомкам. Родитель с этим синдромом имеет 50% вероятность рождения ребёнка с такой же патологией. Это характерно и, для описанных выше синдромах Аперта и Крузона. Различают три варианта этого синдрома. Первый (I) тип считается классическим вариантом синдрома, для него характерно наличие краниостеноза – брахицефалии (в большинстве случаев при данном типе синдромального краниостеноза в патологический процесс вовлекаются коронарный, лямбдовидный и сагитталный швы свода черепа) недоразвитие средней зоны лица, аномалия пальцев рук и ног, сохранный интеллект. Более тяжёлыми считаются варианты IIи III, часто дети рождаются нежизнеспособными и погибают вскоре после рождения (в 25-85% случаях), у них выраженный экзофтальм из-за грубого недоразвития средней зоны лица, тяжелая патология конечностей в виде анкилозов локтевых и коленных суставов, аномалий пальцев рук и ног, грубые неврологические нарушения и задержка интеллектуального развития. Отличие между IIи IIIтипами состоит главным образом в варианте краниостеноза и соответственно, форме черепа. При IIтипе череп имеет форму трилистника (клеверный лист, Kleeblattschädel). Существуют и переходные формы, когда нельзя чётко отнести патологию к какому-либо одному из 3-х вариантов. Аномалии рук и ног, характерные для синдрома проявляютя короткими и широкими большими пальцами рук и ног.
Так же дети с синдромом Пфайффера имеют в 50% случаев проблемы со слухом по причине аномально-малого слухового прохода и среднего уха, зрением по причине маленьких глазниц и аномального расположения структур глазницы, а так же по причине повышенного внутричерепного давления, мальформации Киари. Причём частота встречаемости этих нарушений увеличивается при типах от I до III. Ма
Этот вариант краниостеноза можно диагностировать при внутриутробном УЗИ исследовании, для него характерно наличие экзофтальма, гипертелоризма и широкие большие пальцы рук и ног, определяемые при исследовании.