ГУРЛЕР СИНДРОМ (Пфаундлера – Гурлер болезнь, описана педиатрами – австрийским G. Hurler, 1889–1965, и немецким M. V. Pfaundler, 1872–1947; синонимы – мукополисахаридоз I типа, синдром Шейе, фенотип Гурлер – Шейе) – тяжелое наследственное заболевание; характеризуется недостаточностью альфа-L-идуронидазы – фермента, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Выделяют две аллельные формы синдрома. Первая форма – собственно синдром Гурлер, проявляется в первые месяцы жизни грубыми чертами лица (гаргоилизм), запавшей переносицей, помутнением роговицы, гепатоспленомегалией, тугоподвижностью суставов, деформацией позвоночника; на 2-м году жизни появляются короткая шея, широко расставленные глаза, широкий нос, толстые губы и язык, открытый рот, маленькие редкие зубы; череп нередко имеет форму киля лодки. Типичны помутнение роговицы; тугоподвижность суставов, деформации скелета; макроглоссия, гепатоспленомегалия; клапанные пороки сердца, аномалии коронарных артерий, поражение миокарда по типу гипертрофической кардиомиопатии; задержка роста. Психомоторное развитие до 2 лет, как правило, нормальное, далее отмечается выраженная умственная отсталость, развиваются глухота, слепота. Прогноз неблагоприятный, больные редко доживают до 10 лет. Вторая форма – синдром Шейе (описан американским офтальмологом Н. G. Scheie, 1909–1990), проявляется в школьном возрасте незначительным отставанием в росте, укорочением шеи и туловища, тугоподвижностью суставов, поражением аортального клапана, помутнением роговицы. Течет более доброкачественно, интеллект обычно не страдает. При обследовании выявляют вакуолизированные лимфоциты в мазках крови и повышенную экскрецию мукополисахаридов (дерматансульфат, гепарансульфат) с мочой. Тип наследования – аутосомно-рецессивный. Возможна пренатальная диагностика с помощью исследования обмена мукополисахаридов в культуре клеток амниотической жидкости, получаемой с помощью трансабдоминального амниоцентеза. Лечение включает трансплантацию роговицы, применение диуретиков и вазодилататоров при сердечной недостаточности, антибактериальную профилактику инфекционного эндокардита при поражениях клапанов сердца, при необходимости – их протезирование.


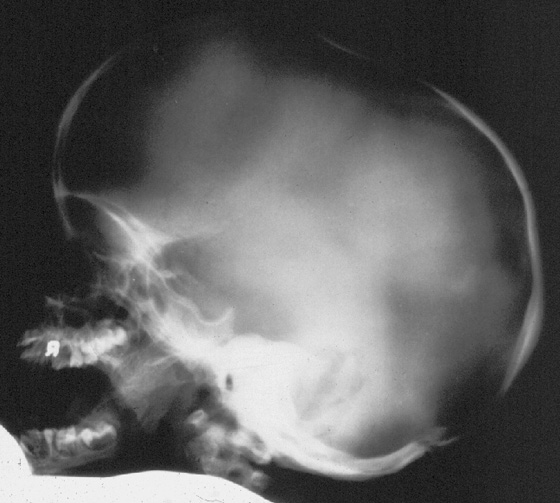
Характерно классическое растягивание в виде J-формы турецкого седла.
Синдром Гурлер (мукополисахаридоз типа I, МПС-I, неправ. назв. – синдром Гурлера) – тяжелое наследственное заболевание обмена веществ.
Синдром Гурлер относится к классу болезней, называемых болезнями накопления. При таких заболеваниях в результате генетического дефекта нарушен синтез какого-либо фермента, необходимого для нормального обмена веществ. В организме накапливаются продукты обмена, которые в норме должны разрушаться этим ферментом. Их избыток вызывает поражение одной или многих систем органов, которое со временем углубляется.
В случае синдрома Гурлер речь идет о дефиците альфа-L-идуронидазы – фермента, необходимого для расщепления особых веществ соединительной ткани – мукополисахаридов. В клетках откладывается их избыток, что постепенно приводит к повреждению различных органов, включая головной мозг, дыхательную систему, глаза, сердце, печень, селезенку, кости, суставы. Чем дольше происходит накопление этих веществ, тем тяжелее состояние больного, и без лечения к возрасту 10-12 лет наступает смерть.
Синдром Гурлер вызван дефектом гена альфа-L-идуронидазы. Существуют и другие мутации того же гена, при которых уровень фермента понижен не настолько сильно. Поэтому выделяют различные варианты мукополисахаридоза типа I. Наиболее тяжелый вариант – синдром Гурлер, наименее тяжелые случаи объединяют под названием синдрома Шейе, а при заболевании промежуточной тяжести говорят о синдроме Гурлер-Шейе. В нашем словаре речь в основном идет именно о синдроме Гурлер, так как этот вариант встречается чаще и протекает тяжелее.
В группу мукополисахаридозов входят и другие болезни. Они различаются по характеру генетического дефекта, клиническим проявлениям и возможностям лечения.
Частота встречаемости, факторы риска
Синдром Гурлер – редкое заболевание, встречающееся с частотой приблизительно 1 случай на 100 тысяч живых новорожденных.
Мукополисахаридоз типа I наследуется по аутосомно-рецесивному типу. Это означает, что больной ребенок может родиться только в случае, если оба родителя являются носителями болезни, имея по одной копии нормального гена и по одной копии «дефектного» гена (такие носители клинически здоровы). Тогда с вероятностью 25% ребенок получит от обоих родителей именно «дефектный» ген, и в этом случае возникает болезнь. Мальчики и девочки болеют одинаково часто.
Если в семье уже были случае рождения детей с синдромом Гурлер, то перед рождением всех последующих детей рекомендуется консультация генетика.
Признаки и симптомы
Дети с синдромом Гурлер при рождении обычно имеют нормальный вес и рост, могут казаться здоровыми и в первые месяцы жизни развиваются нормально. Но уже в возрасте до года, как правило, проявляются первые специфические признаки заболевания. Из-за воздействия болезни на дыхательные пути возникают частые респираторные инфекции. Дети начинают приобретать внешний вид, характерный для синдрома Гурлер: увеличенная голова необычной формы, грубые черты лица, большие лобные бугры, запавшая переносица, широкий кончик носа, толстые губы, приоткрытый рот, увеличенный язык, короткая шея, низкий рост. Пальцы короткие и плохо гнутся, ограничена подвижность также и в других суставах, из-за чего дети начинают ходить позже сверстников и на полусогнутых ногах. Часто у больных диагностируют дисплазию тазобедренных суставов. Грудная клетка деформирована. Живот увеличен из-за большого размера печени и селезенки. Возникают изменения глаз (включая помутнение роговицы), ухудшаются зрение и слух. Со стороны сердечно-сосудистой системы обычны пороки сердечных клапанов.
Характерная черта больных с синдромом Гурлер – отставание в интеллектуальном развитии. Сначала это развитие происходит по возрасту, но уже к 1-1.5 годам замедляется, а к 2-4 годам постепенно прекращается. Затем возникает регресс, то есть утрата уже приобретенных навыков.
Диагностика
Обычно можно довольно рано заподозрить синдром Гурлер по совокупности симптомов и внешних признаков, развивающихся у больного.
Для подтверждения диагноза производится определение уровня альфа-L-идуронидазы в клетках крови (энзимодиагностика) и биохимический анализ мочи. При синдроме Гурлер уровень фермента резко снижен, а в моче обнаруживается существенно повышенное количество мукополисахаридов. Возможен и генетический анализ; при этом не только подтверждается наличие заболевания, но и определяется конкретный генетический дефект.
Применяются также дополнительные обследования: рентгенографическое исследование скелета (при котором обнаруживаются характерные изменения костей), эхокардиография (для обнаружения изменений в сердце), микроскопическое исследование клеток для обнаружения «отложений» мукополисахаридов и т.п.
Семьям, где уже были случаи рождения детей с синдромом Гурлер, может быть предложена пренатальная диагностика при последующих беременностях.