Синдром Протея - редкое врождённое заболевание. СП впервые был описан Коэном и Хайдом в 1979 году, когда этот синдром был выявлен у 2 пациентов [3]. Позже, немецкий педиатр Ганс-Рудольф Видеман назвал это заболевание синдромом Протея, в честь греческого бога Протея "полиморфного" из-за его изменчивых форм [11]. СП - комплексное гамартомное заболевание, характеризующееся чрезмерным разрастанием тканей (макродактилия или гемигипертрофия), подкожными опухолями, разными костными и кожными аномалиями [4]. Возможно, эти характеристики являются определяющими при постановке диагноза. Диагностировать СП сложно, так как клинические проявления болезни совпадают с другими аномалиями тканей и гамартоматомными расстройствами [7]. Характерный диагноз включает в себя различные заболевания, связанные с разрастанием тканей - костей, кожи и центральной нервной системы. Макродактилия может быть общей чертой и других болезней тоже. СП следует отличать от других заболеваний, таких как гамартозы, поражения кожи, сосудистые аномалии и нервно-кожные заболевания. При постановке диагноза СП, всегда следует помнить, что при СП разрастание тканей носит асимметричный характер.
![]()

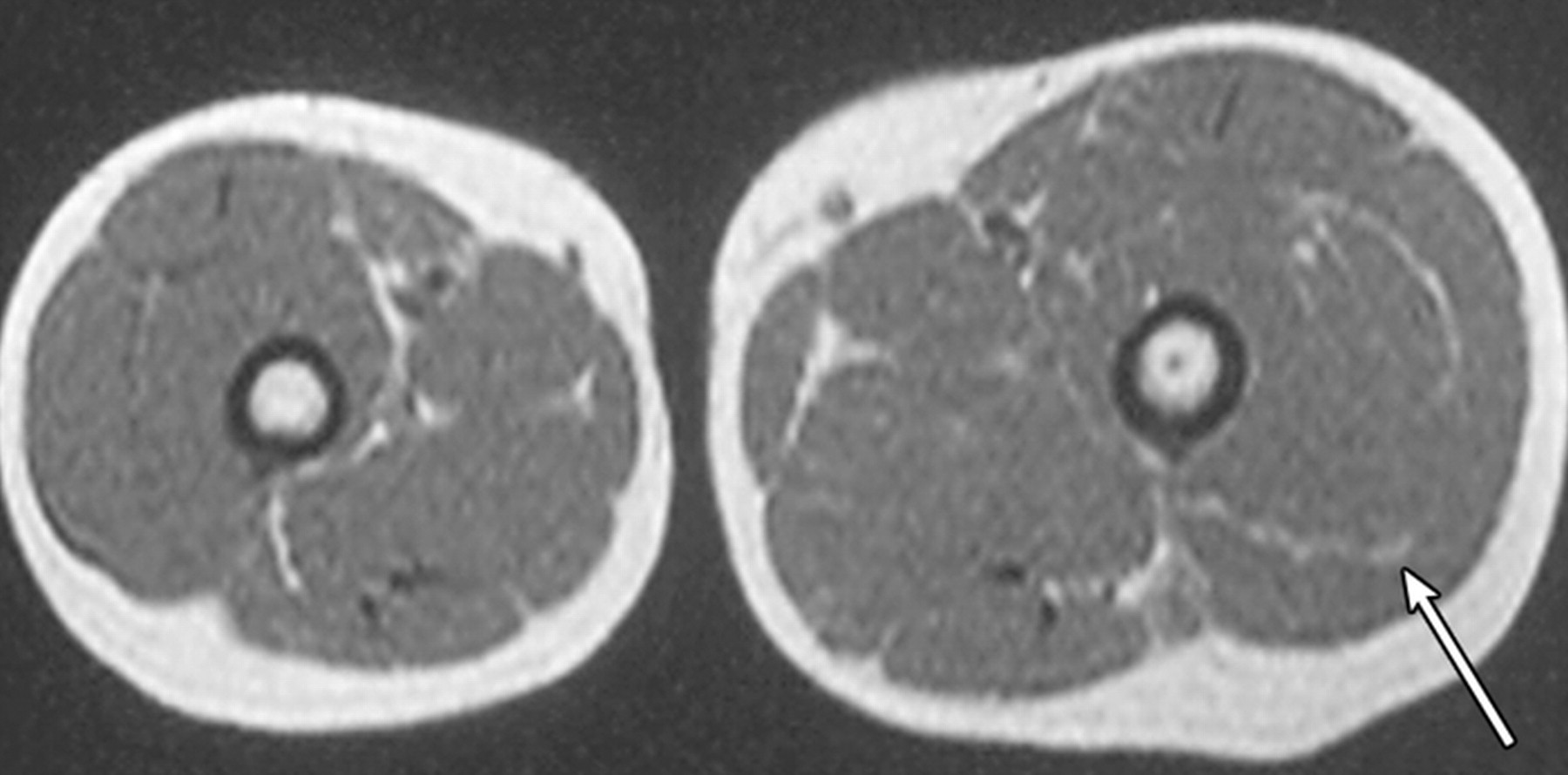

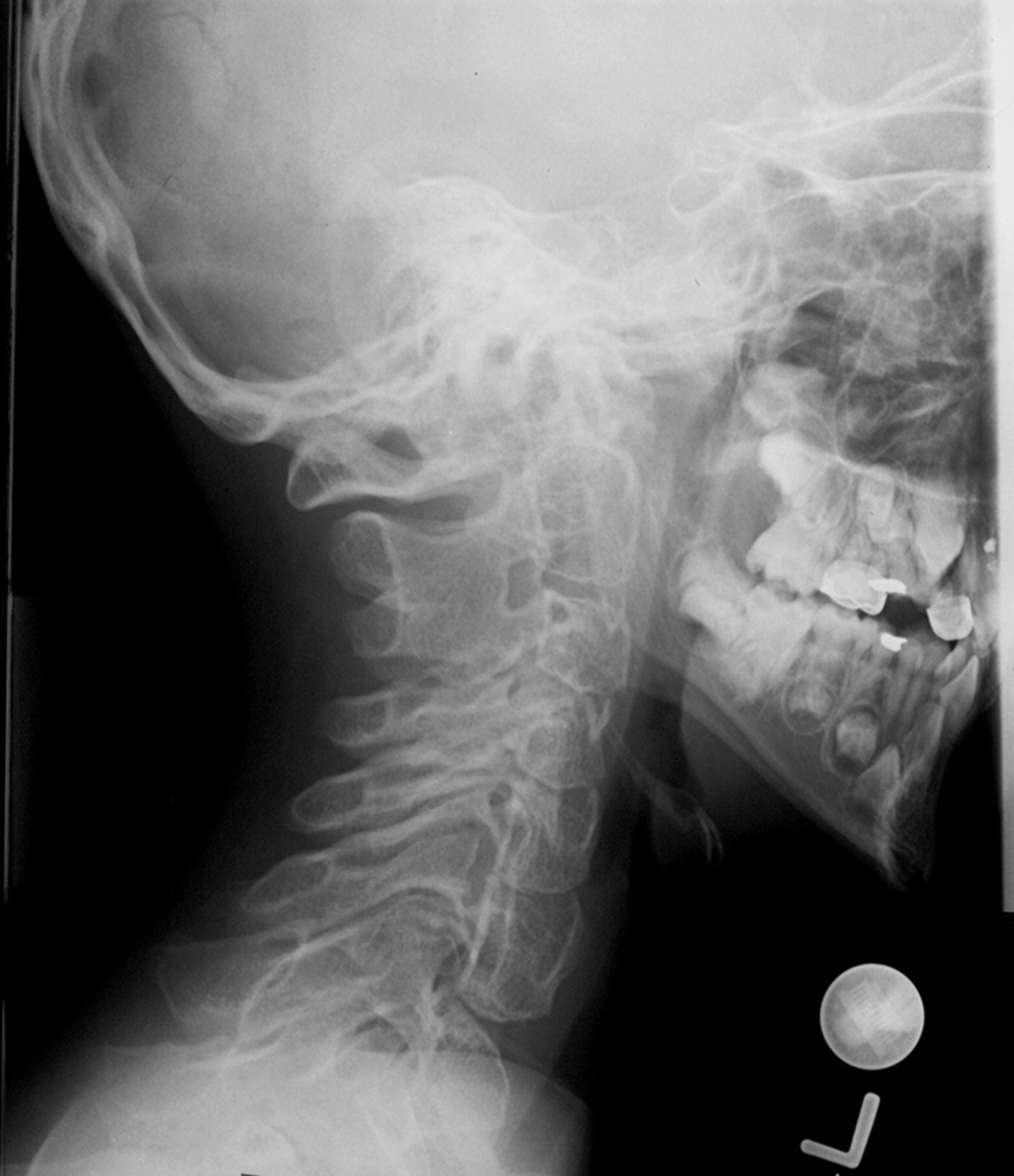

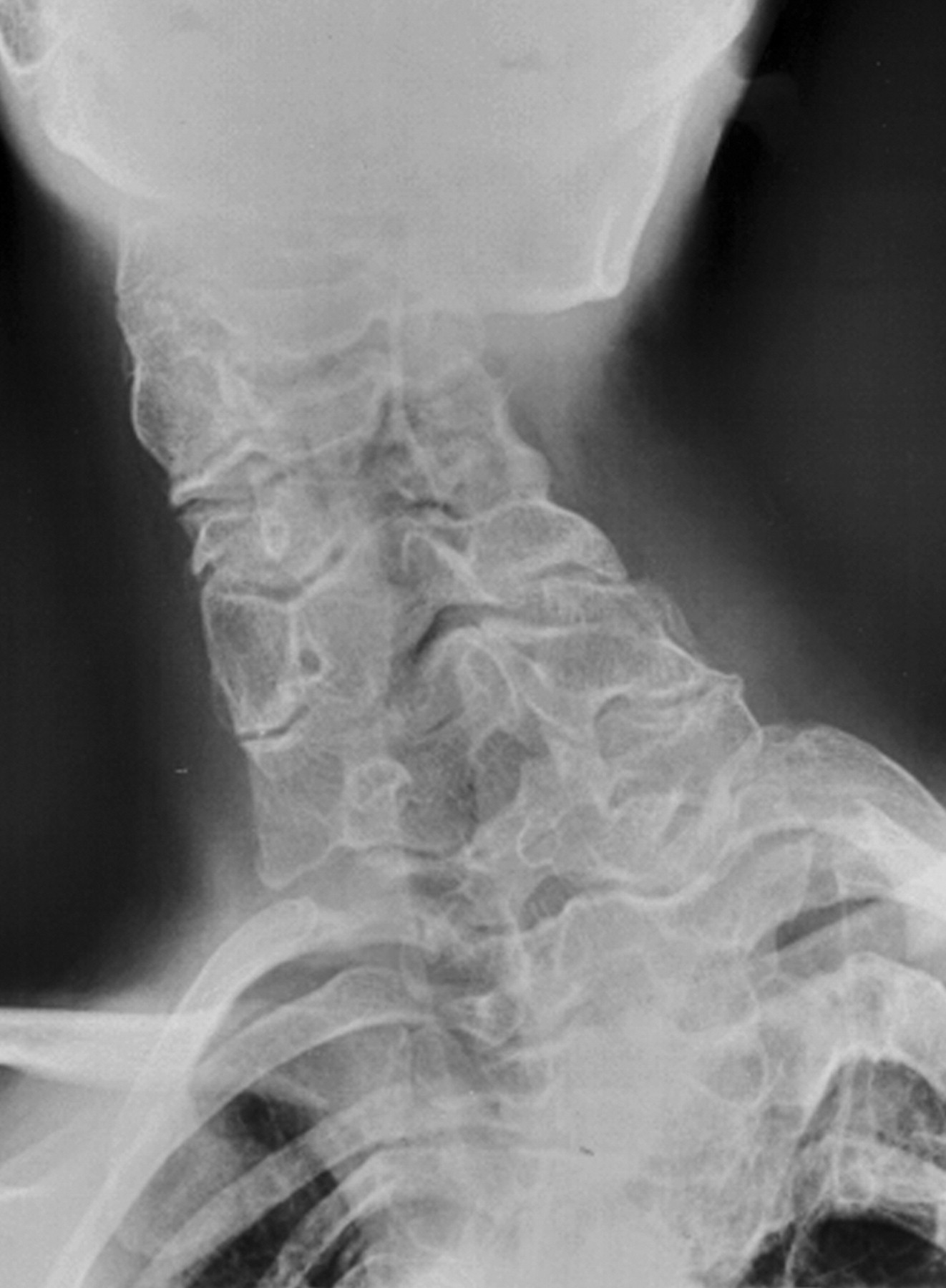
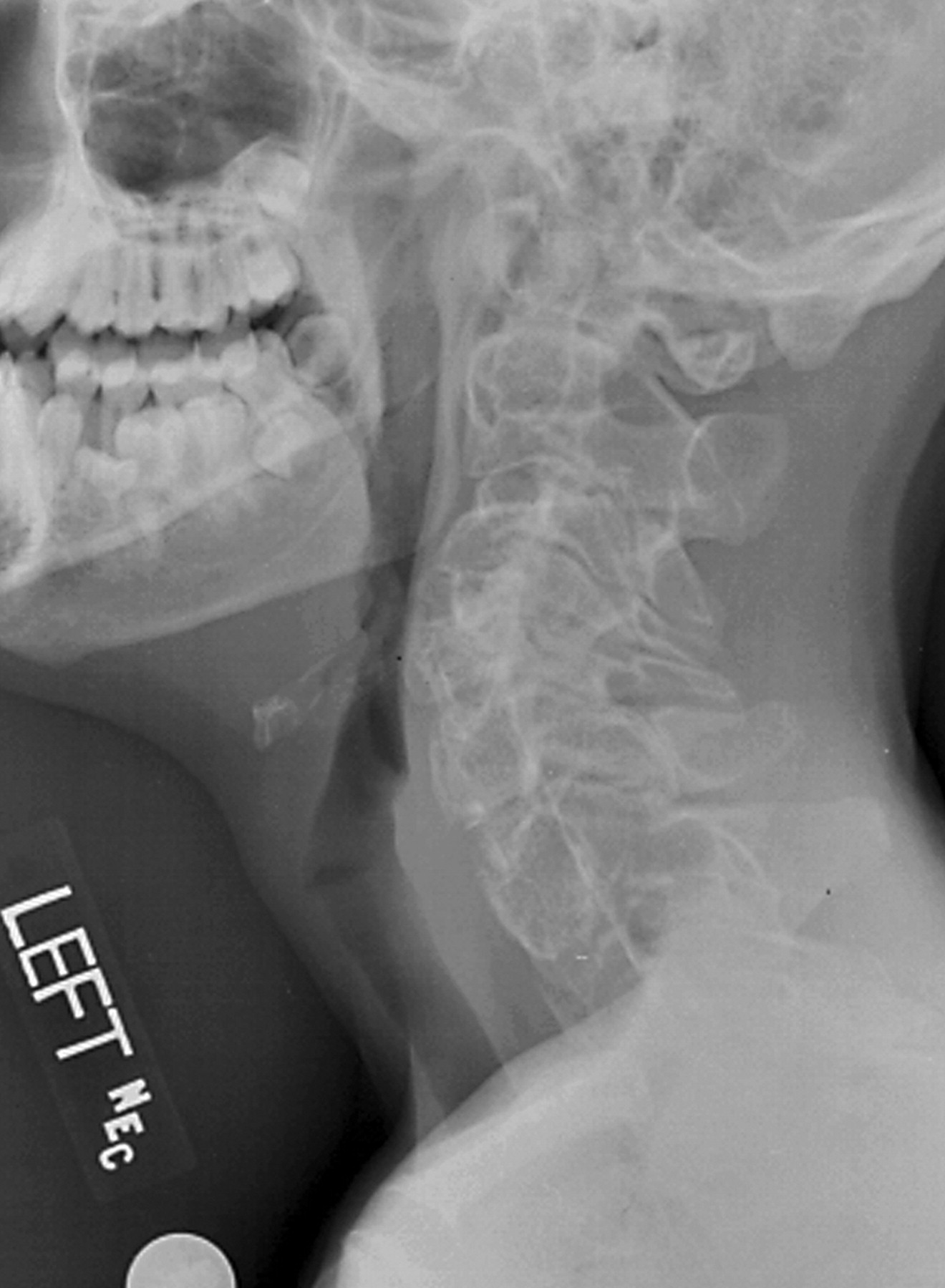

http://www.radiomed.ru/forum/material-ryadom-no-material-interesnyi?page=1
А. Н. Семячкина П. В. Новиков В. Ю. Воинова М. Б. Курбатов Т. А. Синельщикова Н. С. Кузьмина Г. Д. Засухина
Опираясь на данные литературы о синдроме Протея у детей, приведено собственное клиническое наблюдение и диагностическая тактика верификации этого редкого наследственного заболевания. Особое внимание уделено вопросам патогенеза развития основной клинической симптоматики болезни (высокие показатели физического развития и склонность к формированию злокачественных новообразований): парадоксальной реакции гормона роста на стандартный глюкозотолерантный тест и нарушению репаративного синтеза ДНК. Показана сложность дифференциального диагноза синдрома Протея с другими фенотипически сходными патологическими состояниями.
В педиатрической практике чаще встречаются заболевания, сопровождающиеся задержкой роста детей, однако нередко наблюдаются состояния, характеризующиеся увеличением антропометрических параметров больных. Группа этих заболеваний генетически гетерогенна, включает болезни хромосомного, моногенного и полигенного происхождения, которые у педиатров вызывают диагностические трудности. Довольно часто одним из кардинальных симптомов этой патологии является также задержка психоречевого развития детей. В последние годы заметно возросло число сообщений, свидетельствующих о склонности у таких больных к развитию злокачественных новообразований, которые могут иметь самую различную локализацию.
Знание генеза и симптоматики различных синдромов высокого роста у детей определяет целесообразность и эффективность назначения комплекса терапевтических мероприятий, своевременной диагностики и профилактики злокачественных новообразований, а также консультирования семей. На примере синдрома Протея нами разработана тактика дифференциальной диагностики и верификации патологии у детей с высокими антропометрическими показателями.
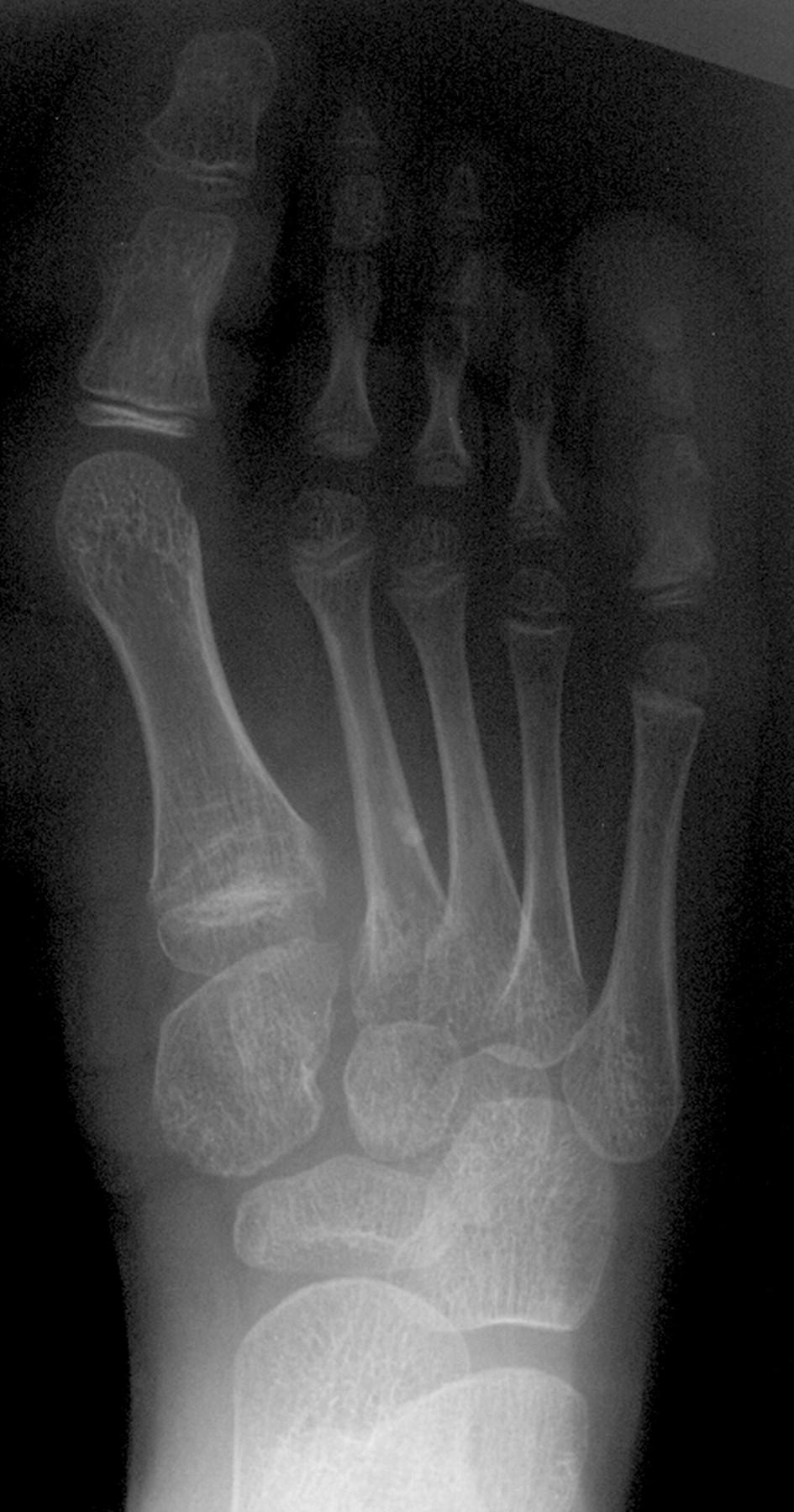
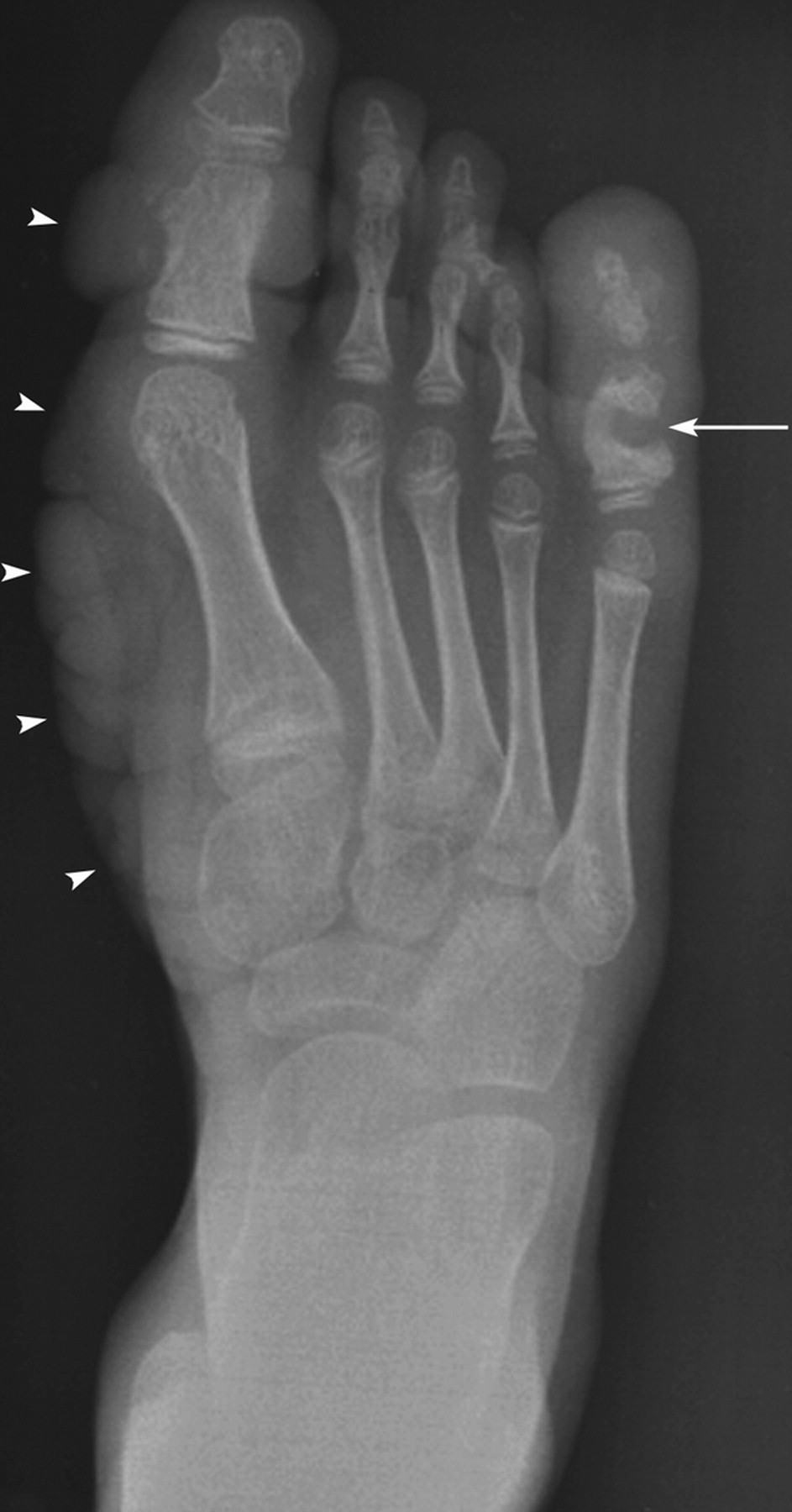
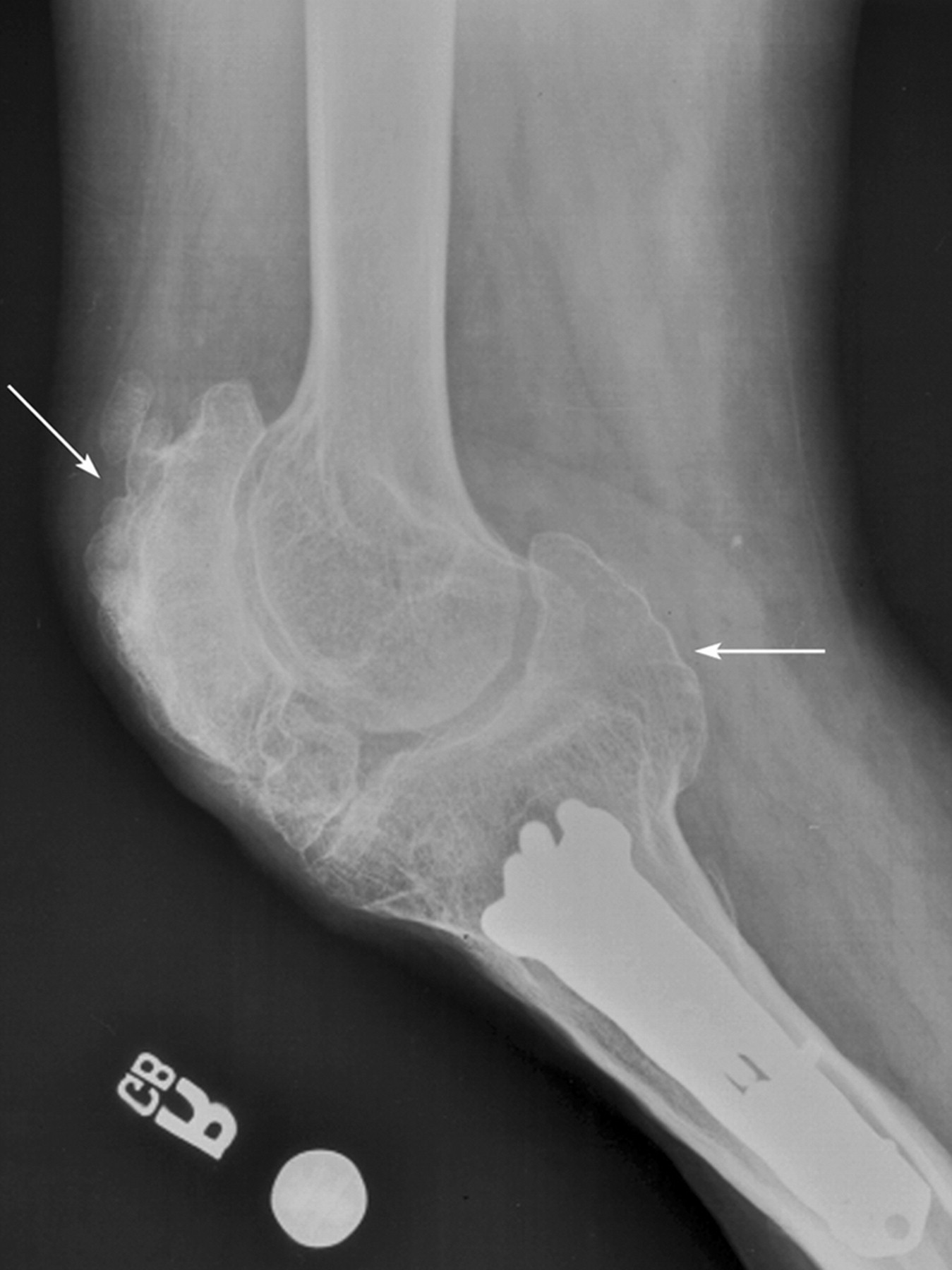
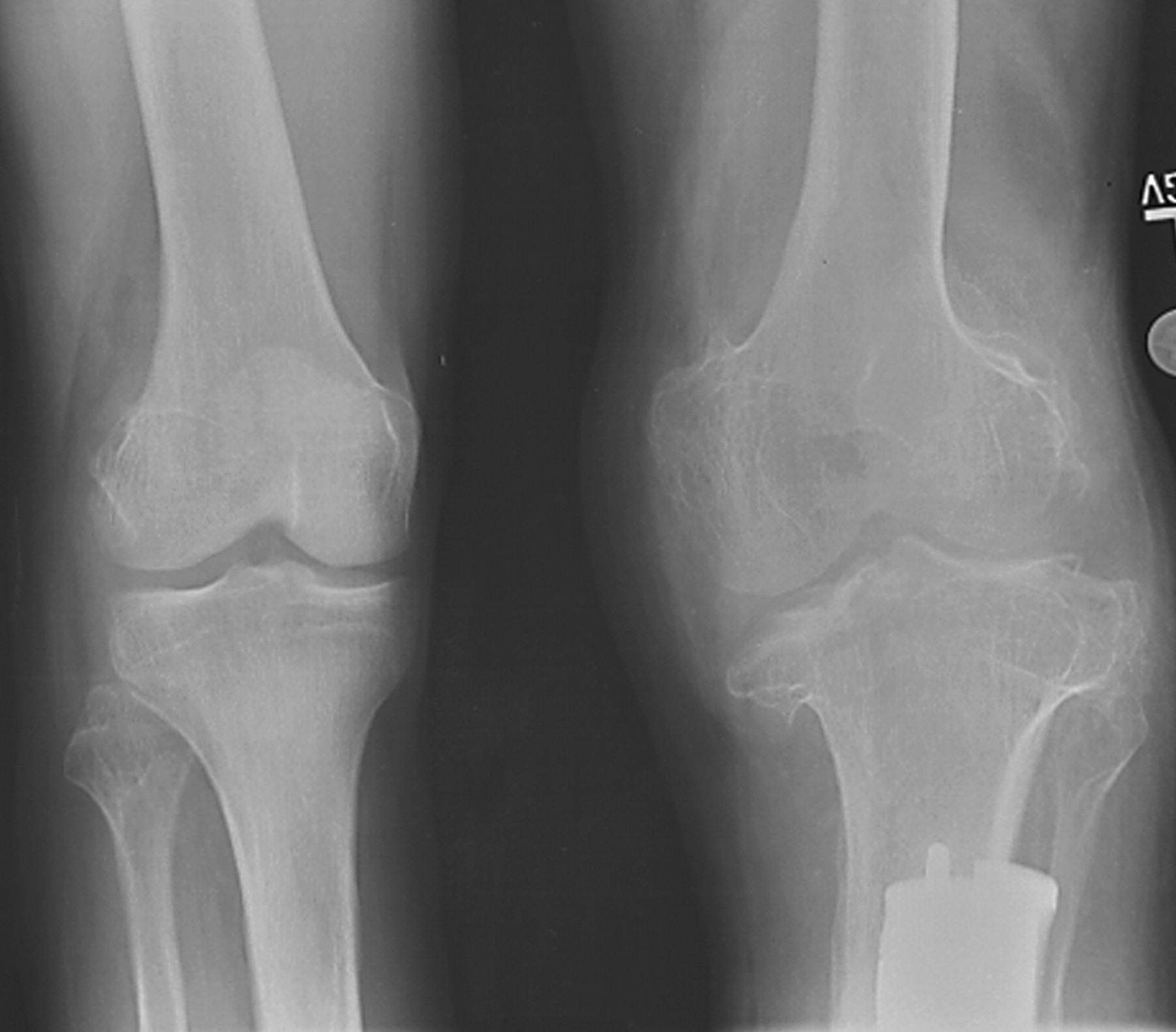
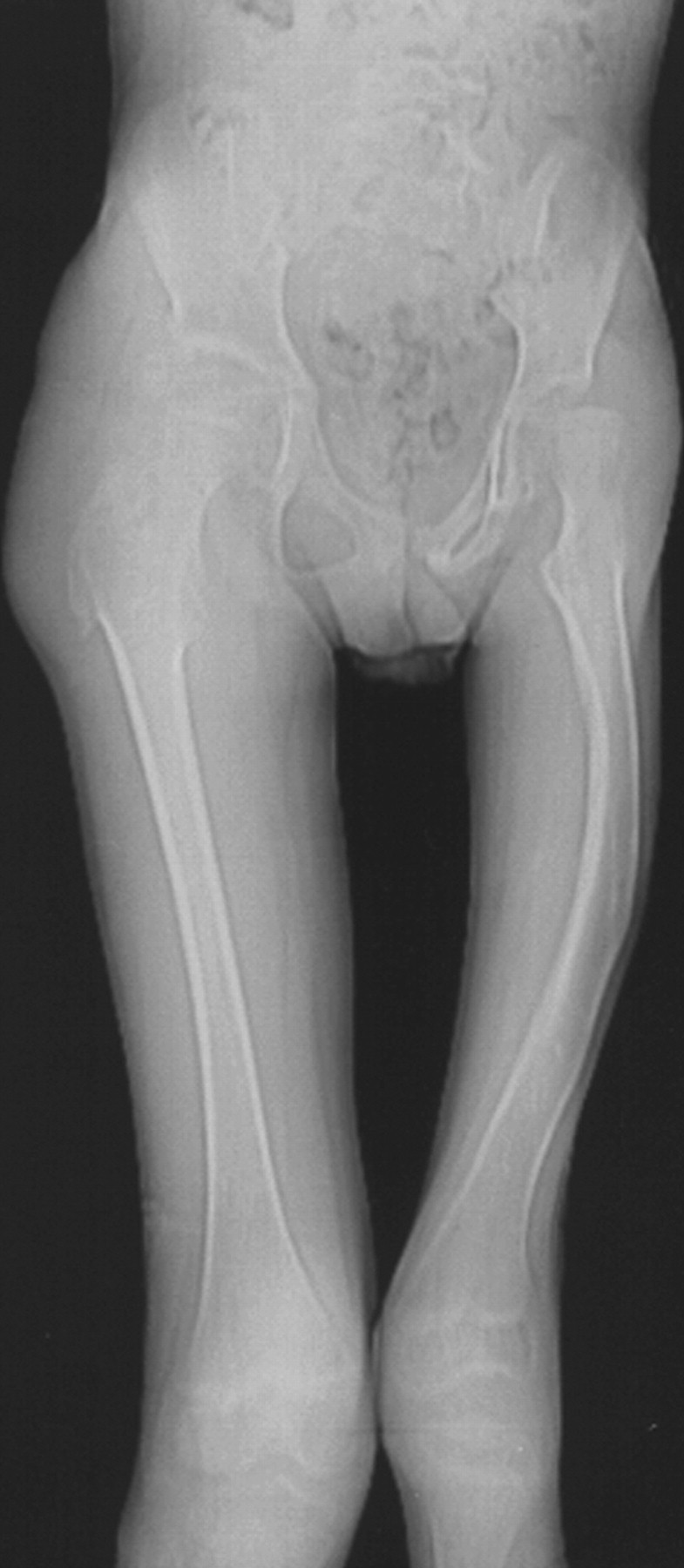
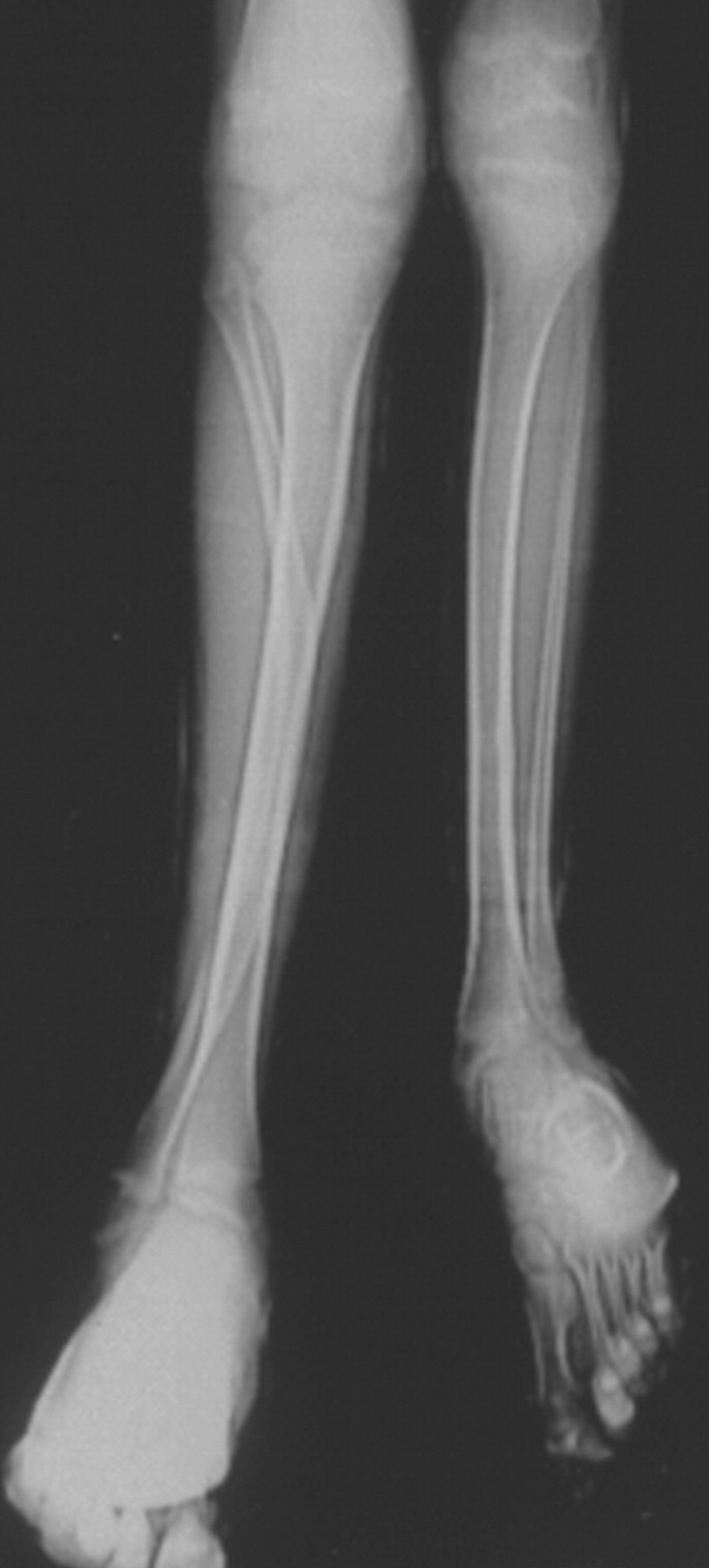
Синдром Протея был описан H. Wiedemann и соавт. в 1983 г. Исследователи сообщили о 4 мальчиках, не состоявших в кровном родстве, у которых имелась сочетанная и идентичная клиническая симптоматика в виде парциального гигантизма рук и ног, невусов, гемипертрофии, подкожных опухолей, макроцефалии, других аномалий черепа, ускоренного роста и висцеральной патологии [1]. Авторы включили заболевание в группу врожденных гамартом генетического происхождения с вероятным аутосомно-доминантным типом наследования. H. Wiedemann и соавт. назвали данную патологию по имени древнегреческого бога Протея (греч. Proteus - «полиморфный»), обладавшего уникальной способностью радикально изменять свой внешний облик с целью более успешного захвата добычи. Исследователи также обратили внимание на тот факт, что заболевание может быть неправильно диагностировано как синдромы Клиппеля-Треноне-Вебера, Маффучи или болезнь Оллье [1].
Несколько позже было установлено, что синдром Протея в действительности впервые был описан M. Cohen и P. Hayden в 1979 г., а H. Wiedemann и соавт. сделали эту болезнь широко известной [2].
Относительно генеза болезни к настоящему времени нет единого мнения. Работы в этом направлении активно продолжаются. Чаще синдром Протея описывается как спорадическое врожденное заболевание. Наряду с этим имеются сведения, указывающие на семейные случаи. Так, J. Goodship и соавт. в 1991 г. сообщили о возможности передачи синдрома Протея от отца к сыну [3].
В 1993 г. G. Kruger и соавт. наблюдали легкую форму синдрома Протея у матери и ее сына [4]. При этом отмечалось, что проявления болезни у женщины были минимальными.
M. Cohen в 1993 г. на основании анализа клинической симптоматики двух случаев синдрома Протея высказал предположение о соматическом мозаицизме [5]. E. Smeets и соавт. в 1994 г. сообщили о больном с локальными проявлениями синдрома Протея, что позволило авторам поддержать гипотезу о роли соматического мозаицизма в генезе данной патологии [6].
В последние годы увеличилось число исследований, посвященных ДНК-диагностике синдрома Протея. Так, X. Zhou и соавт. в 2000 г. [7] опубликовали сообщение о мальчике с врожденной гемигипертрофией, эпидермальным невусом, макроцефалией, липомами, артериовенозными аномалиями и нормальным интеллектом. ДНК-диагностика лимфоцитов периферической крови пробанда, тканей невуса, липомы, артериальных и венозных сосудов выявила две различные мутации в 5-м экзоне гена PTEN (phosphatase and tensin homolog) - гена супрессора опухолей. Установлена локализация гена PTEN на длинном плече хромосомы 10, в локусе 10q23.31. Однако окончательная роль мутаций гена PTEN в реализации симптоматики синдрома Протея не установлена, так как мутации в этом гене найдены еще при ряде других заболеваний, характеризующихся развитием злокачественных новообразований, - синдромах Коудена и Банаян-Райли-Рувалькаба. Так, только у 5 (из 21 обследованного) больных с синдромом Протея были выявлены мутации гена PTEN, что составляет около 20%, в то время как мутации этого же гена зарегистрированы у 80% пробандов, страдающих синдромом Коудена [8]. Работы в этом направлении активно продолжаются.
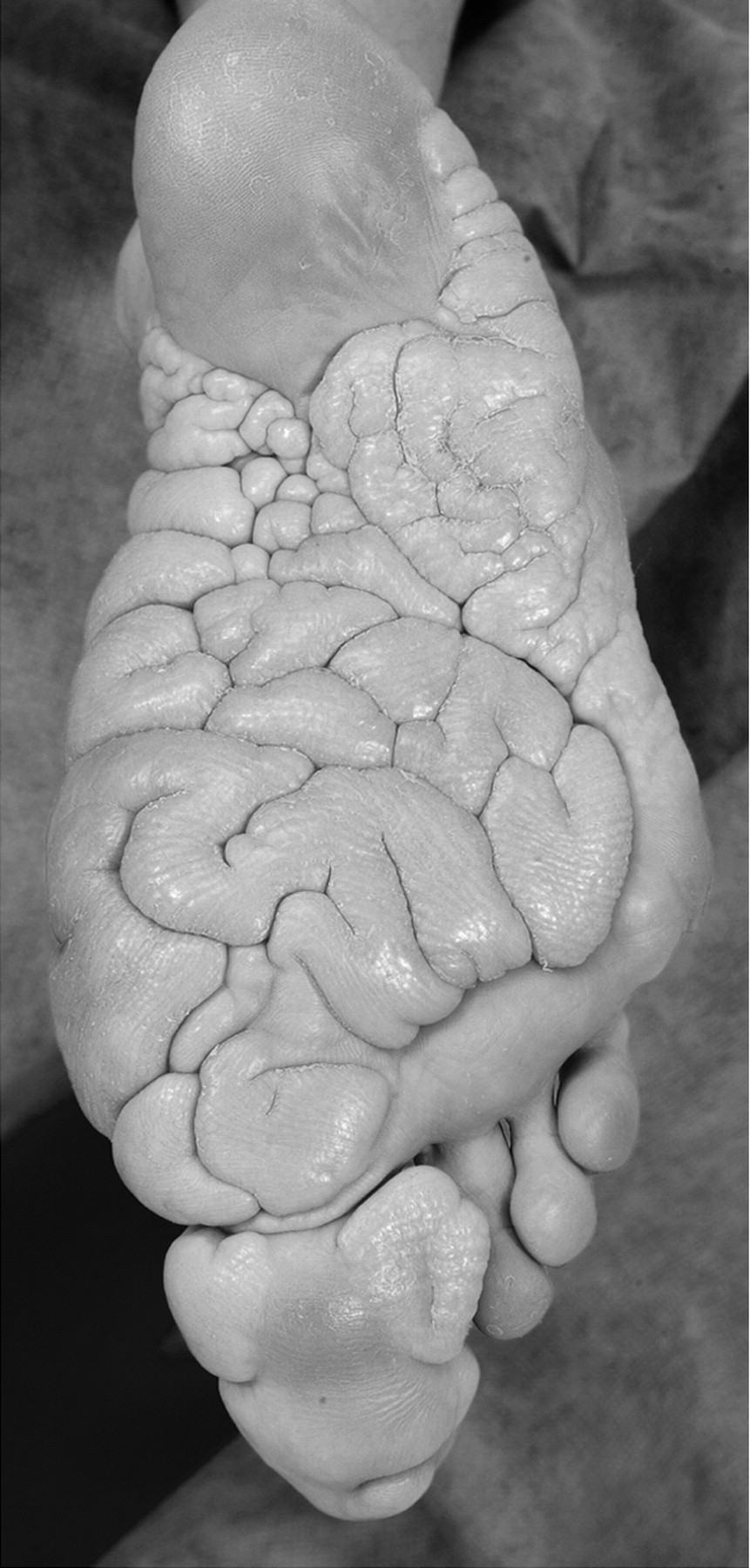
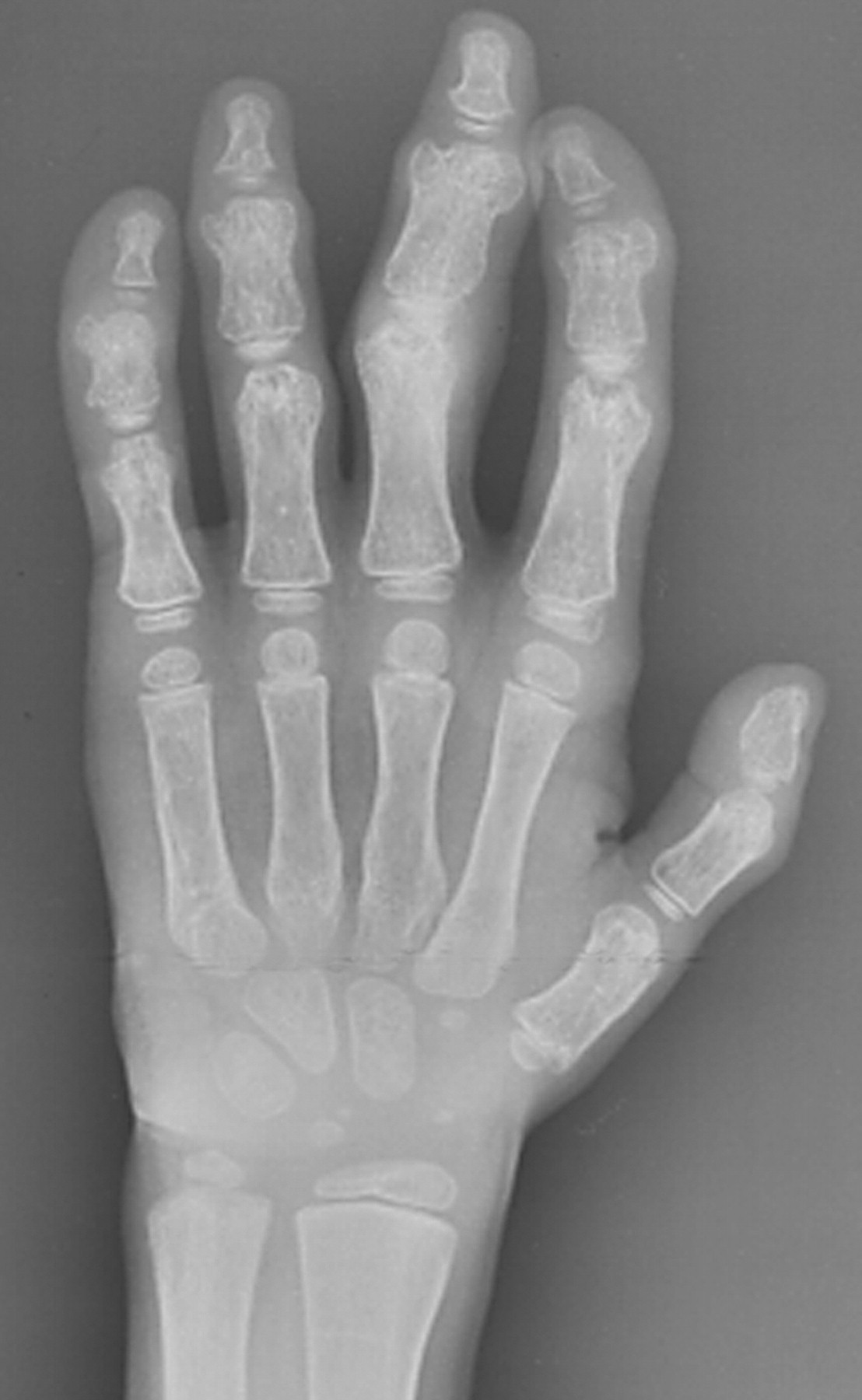
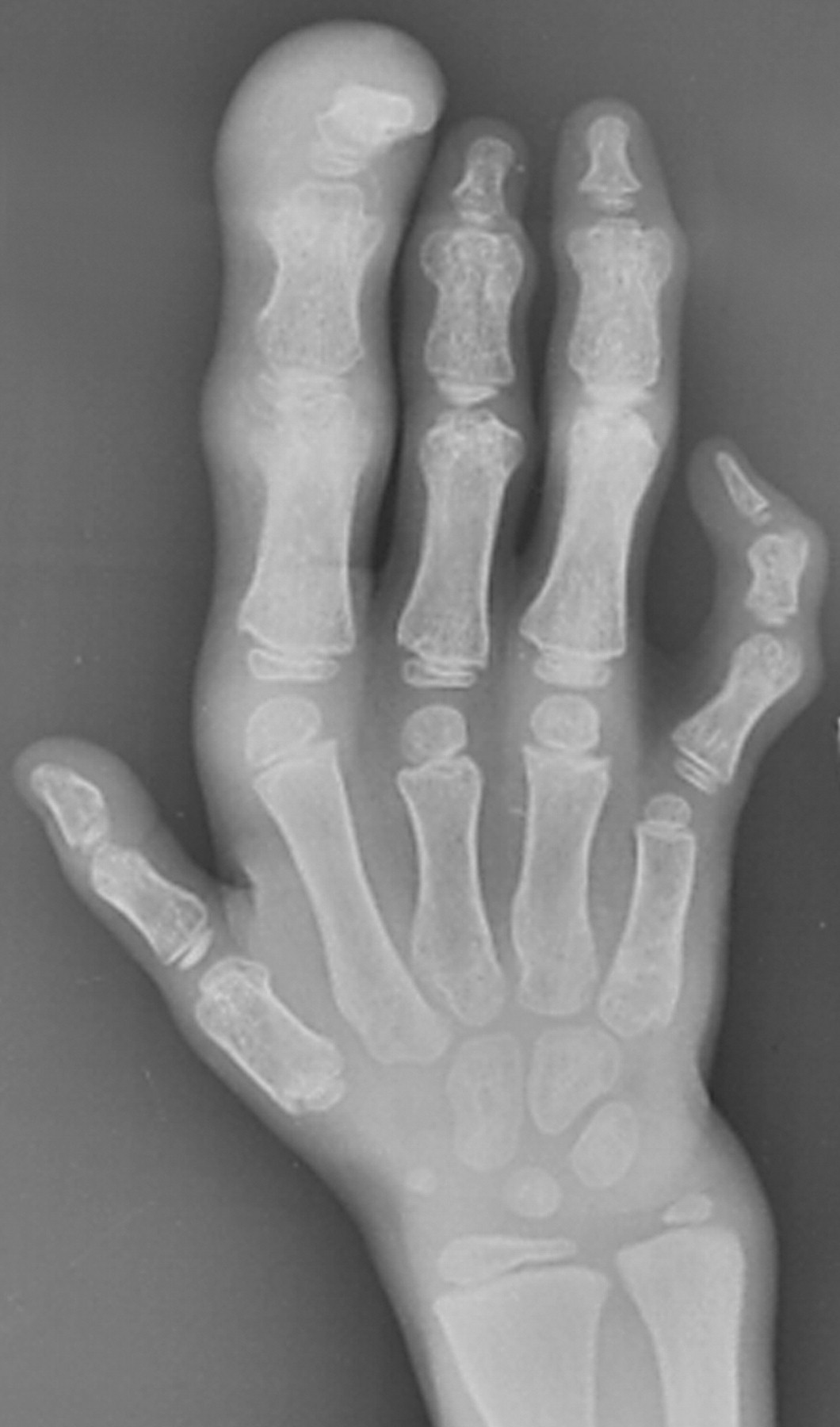
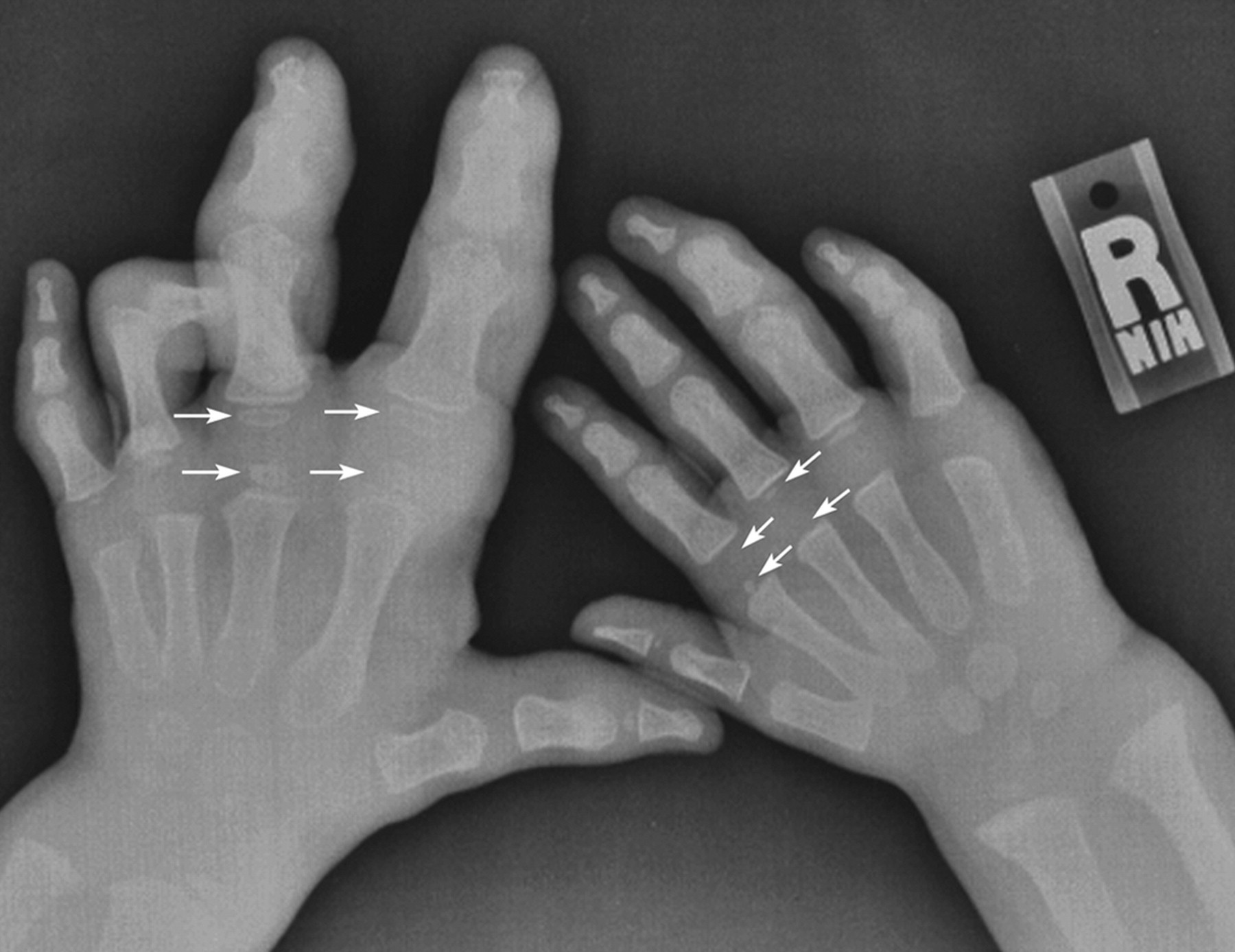
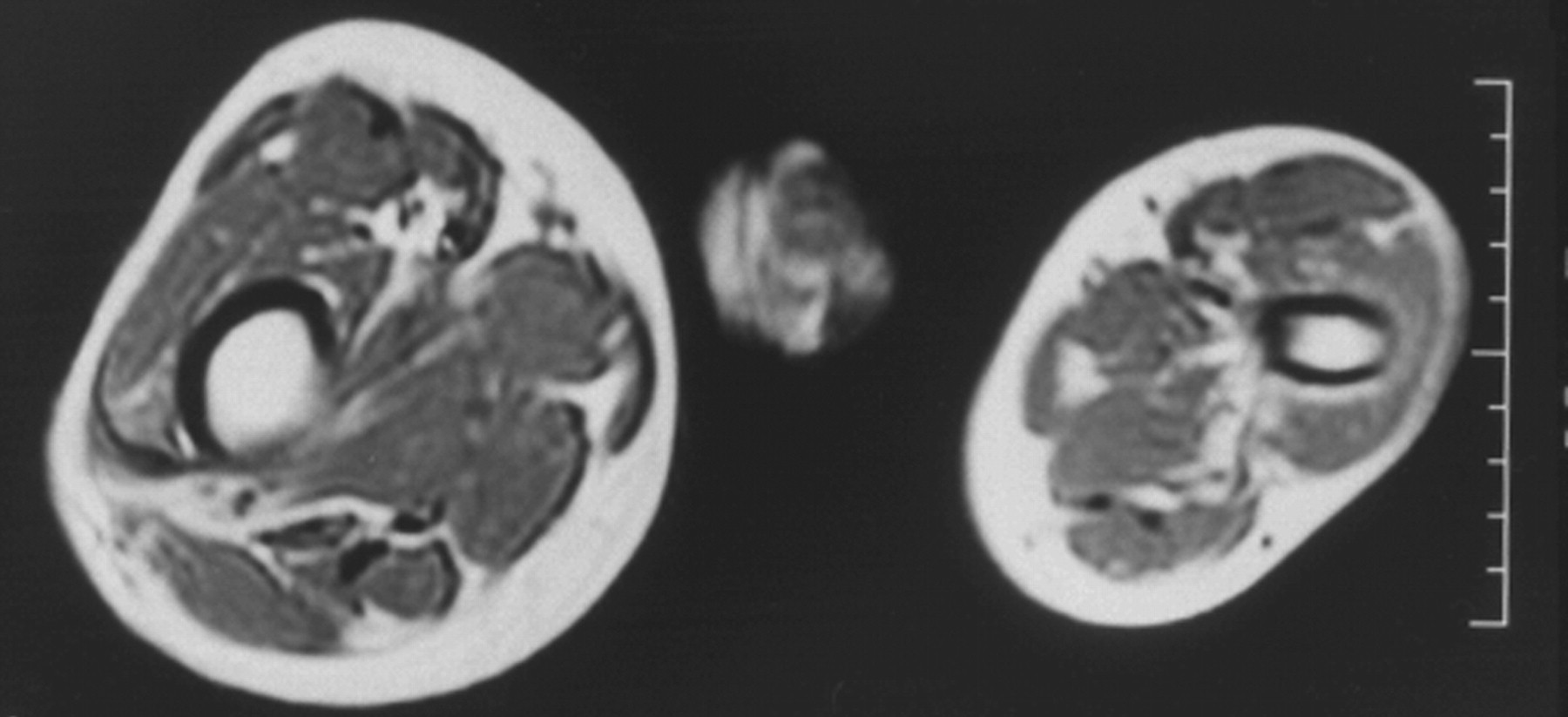
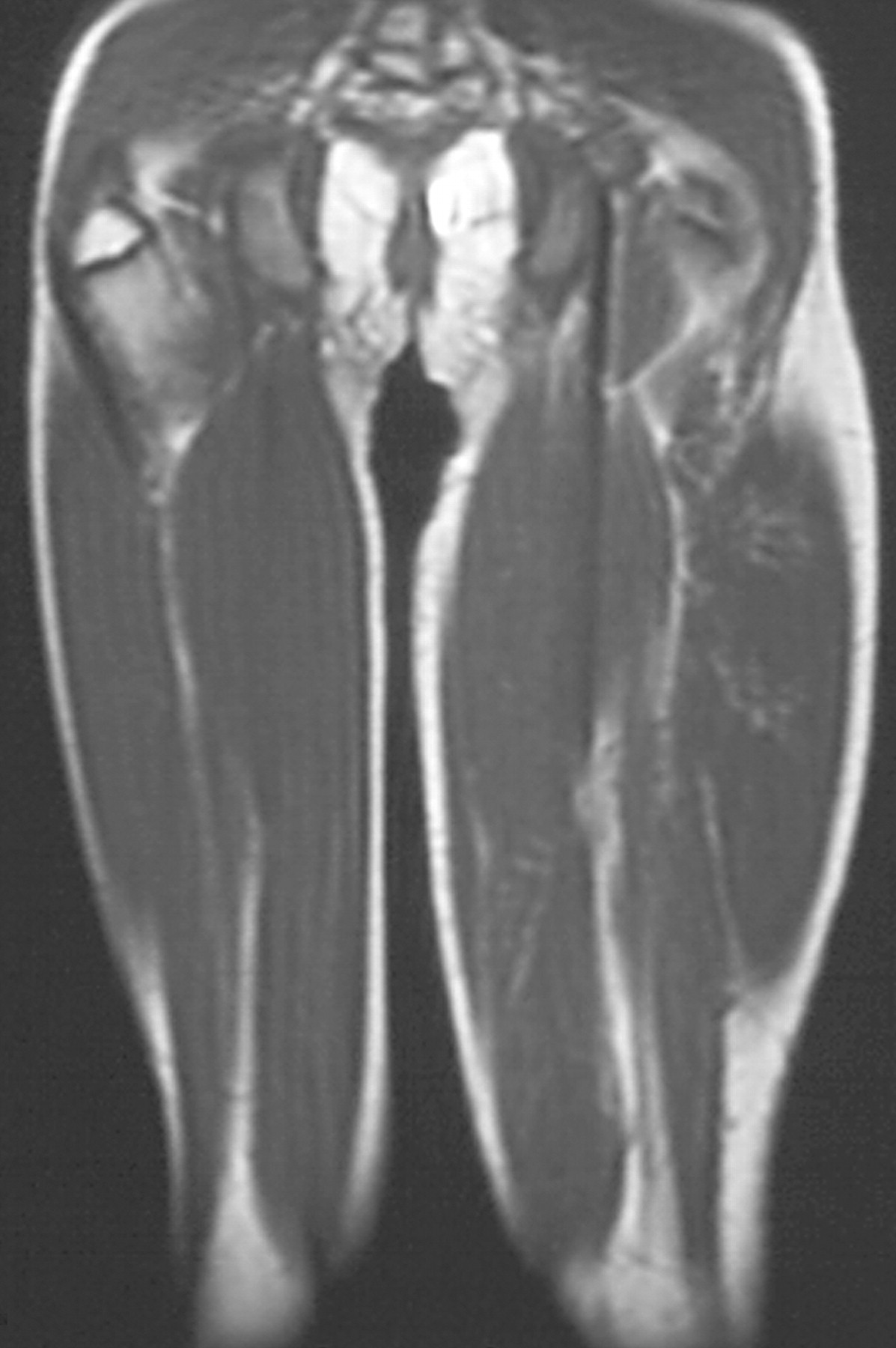
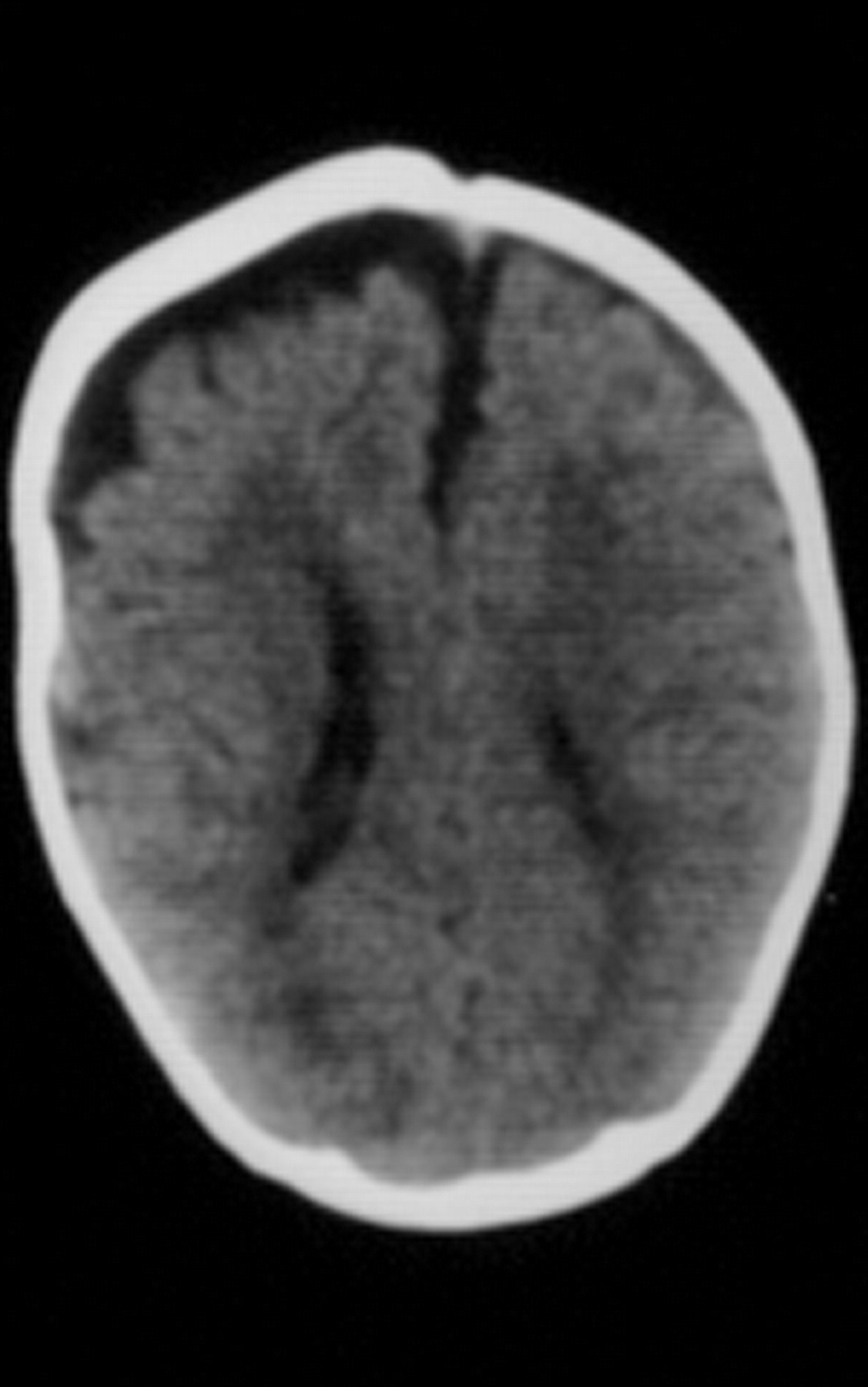
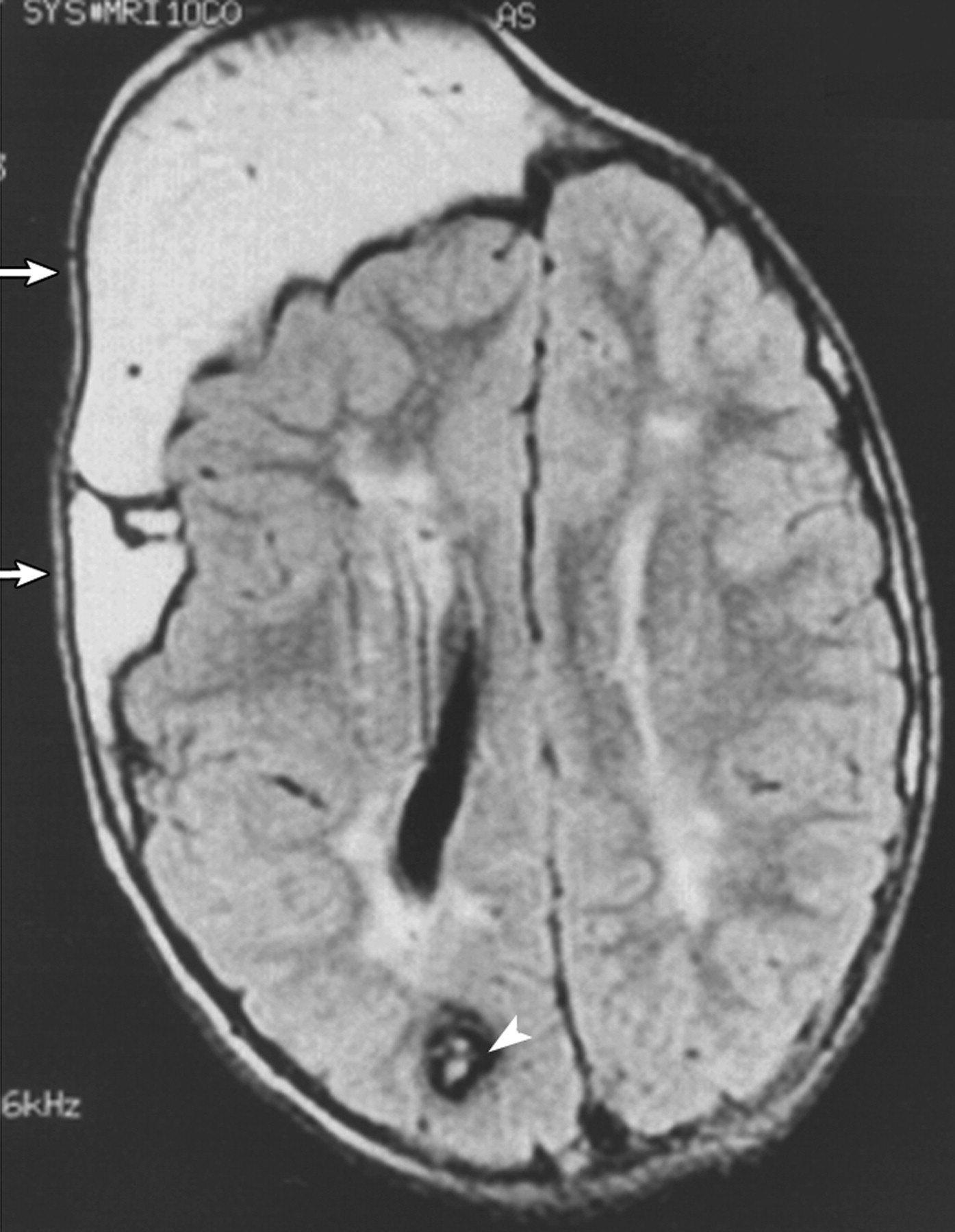
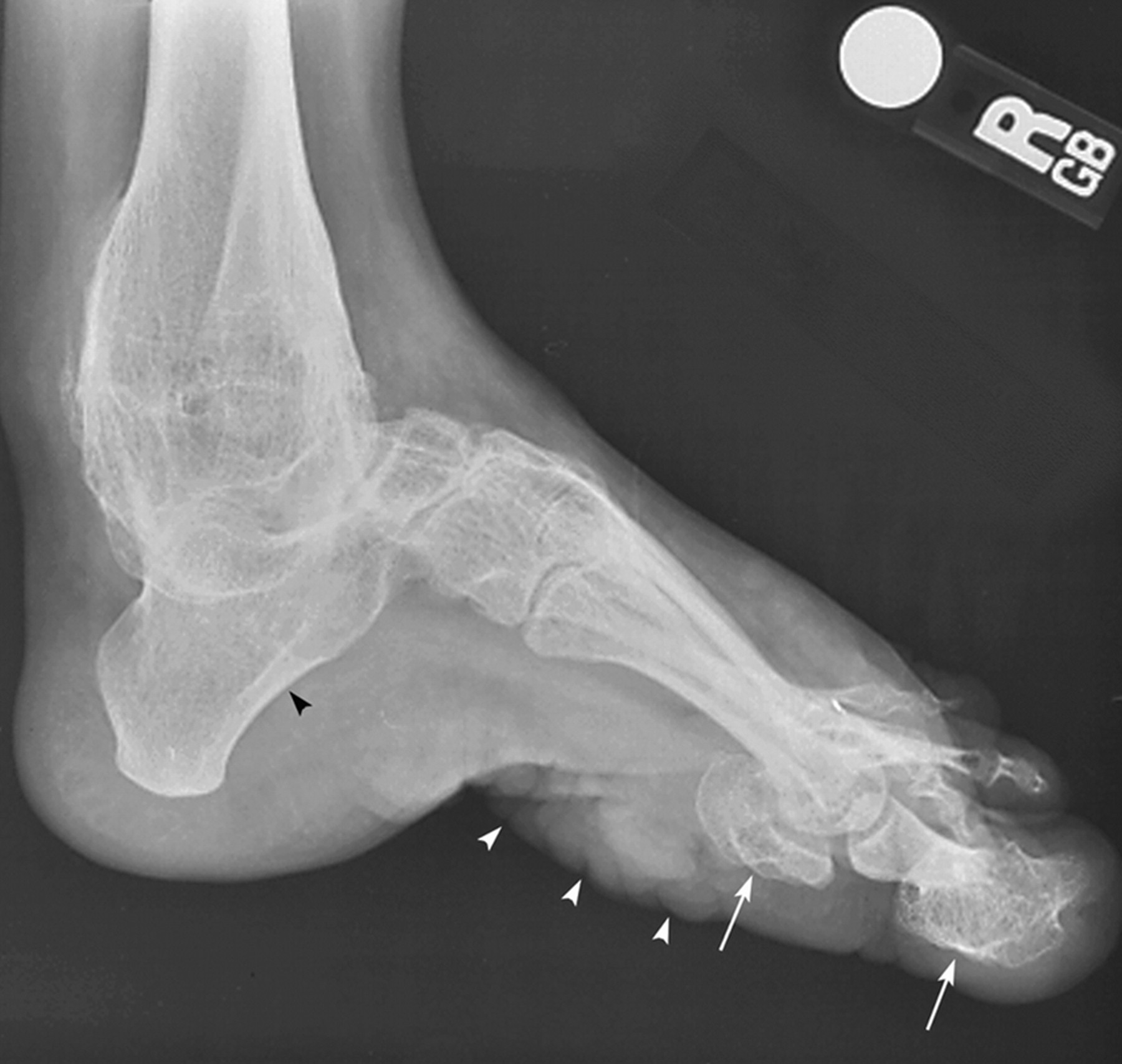
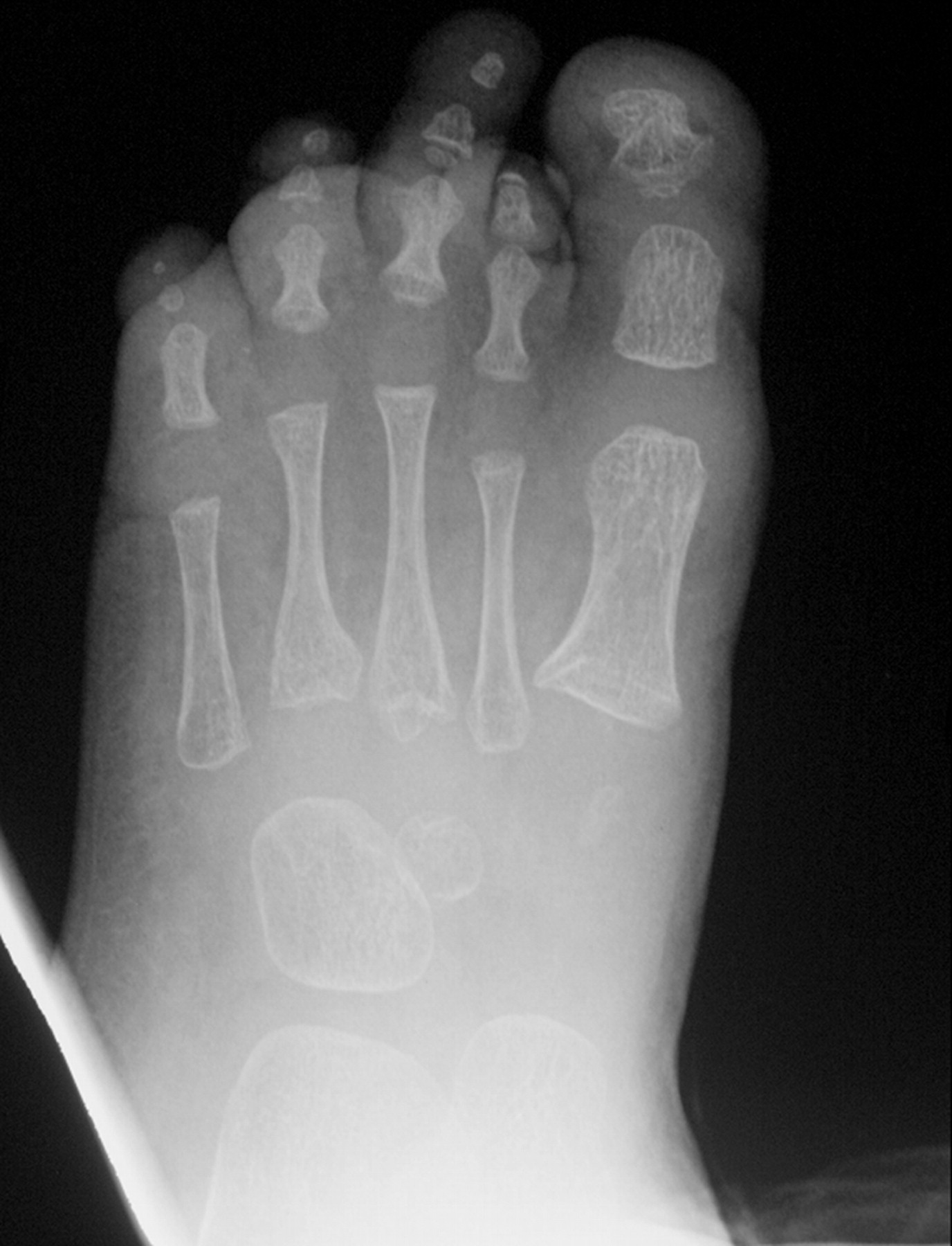
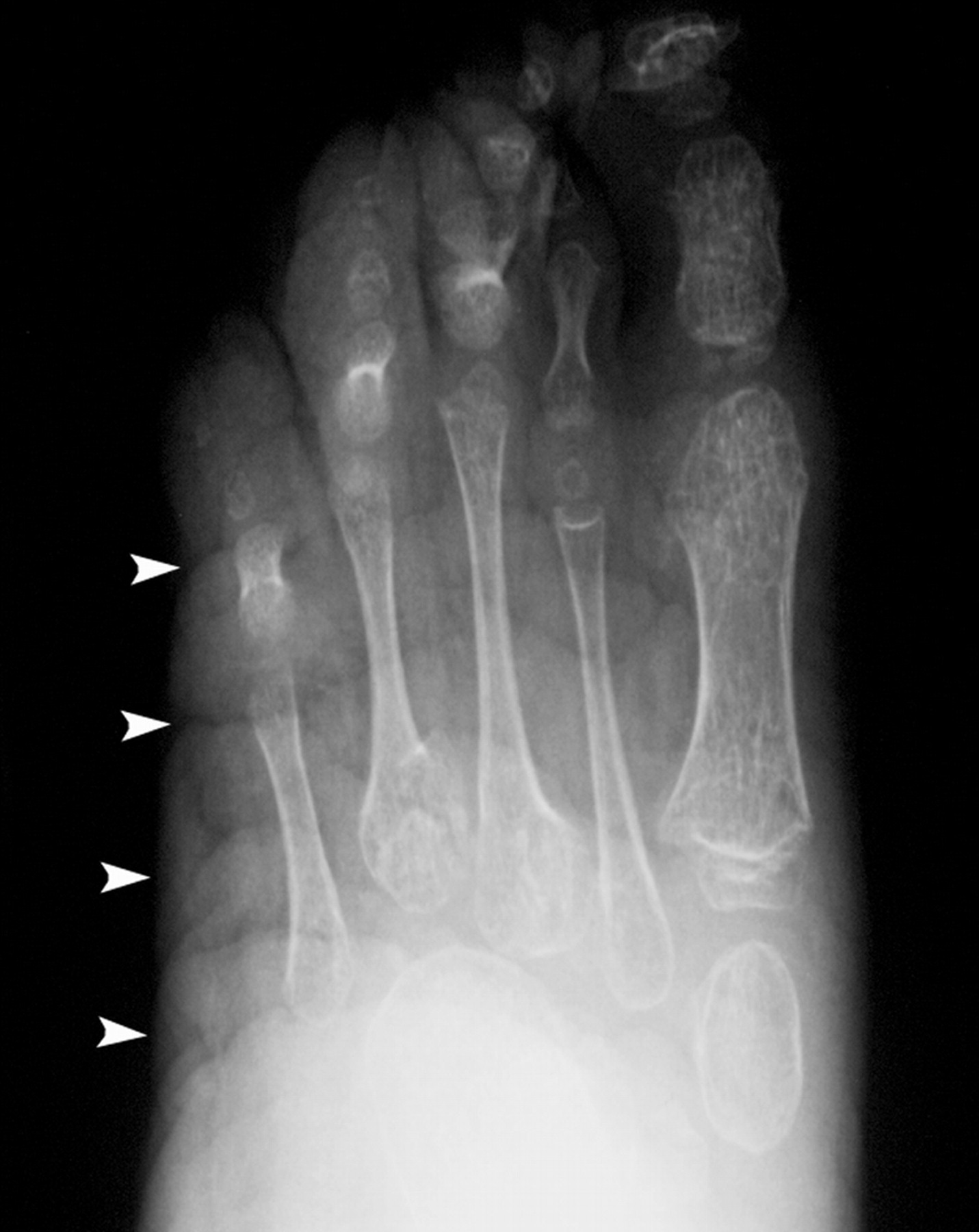
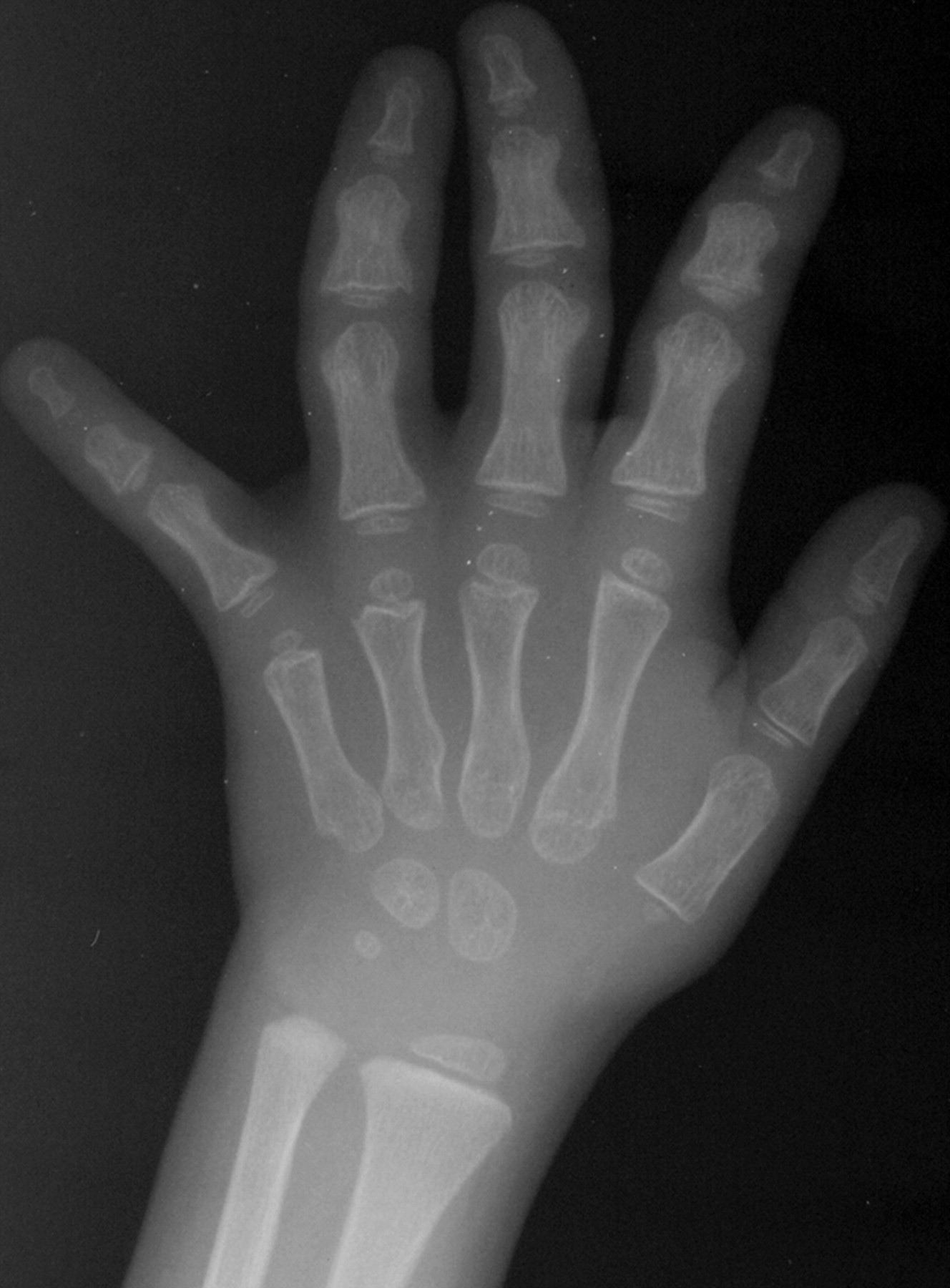
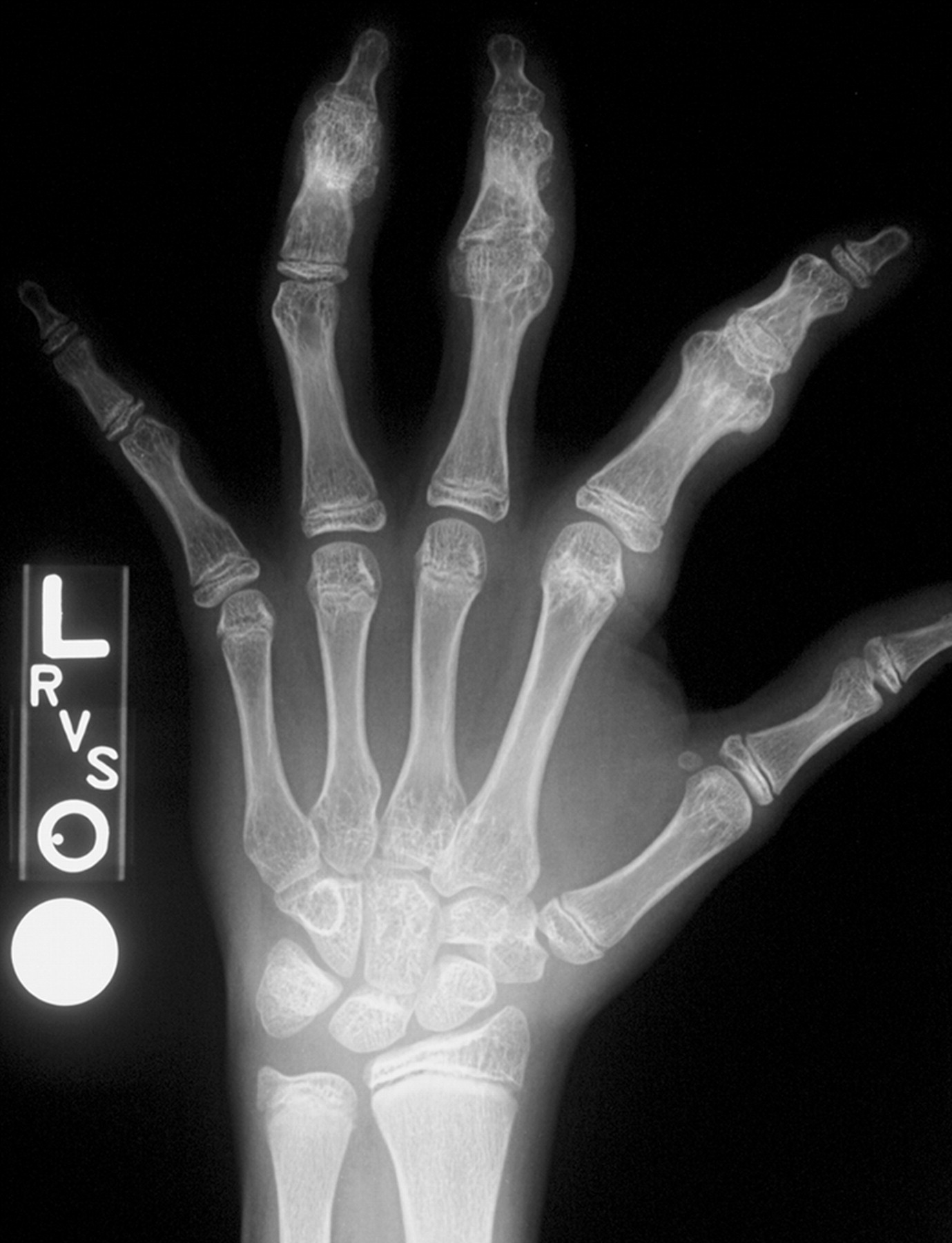
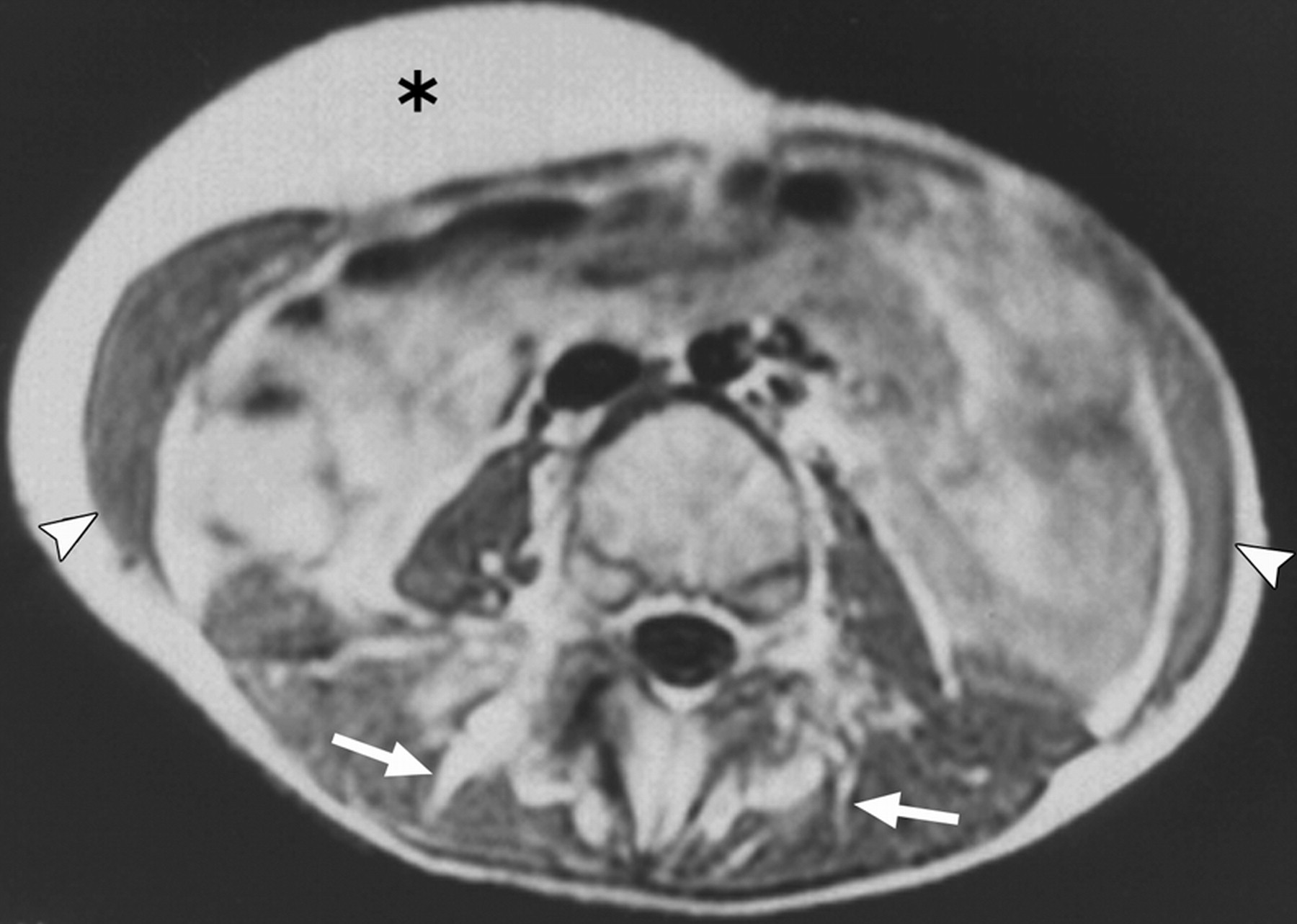
Клинические проявления заболевания характеризуются выраженным полиморфизмом. Наряду с типичной симптоматикой - парциальным гигантизмом рук и ног, гемигипертрофией, артериовенозными аномалиями, макроцефалией - наблюдаются разнообразные опухоли: бородавчатый эпидермальный невус, гемангиомы, лимфангиомы, липомы, гамартомы. Иногда отмечаются косоглазие, экзофтальм, миопия, прогения, варикозное расширение вен, разрастание кожи на подошвах. Примерно в 55% случаев выявляется умственная отсталость, в 13% - судорожный синдром. Наблюдались также больные с изолированной макродактилией, спленомегалией, избирательной патологией глаз и черепа в виде множественных менингиом, полимикрогирии, ретинальной пигментной дегенерации и атрофии зрительного нерва. Продолжительность жизни пробандов, как правило, невелика и колеблется от 3 до 40 лет. Смерть наступает чаще от злокачественных новообразований. Описаны такие опасные для жизни осложнения, как венозные тромбозы и эмболия легочной артерии. Так, A. Stavotinek и соавт. в 2000 г. сообщили о 3 больных, умерших в возрасте 12, 17 и 25 лет от эмболии легочной артерии [9].
A. Mohamedbhai и соавт. описали синдром Протея у новорожденного мальчика, мать которого в 6 нед гестации принимала простагландин с целью прерывания беременности [10].
Протея синдром
http://www.indeedopedia.com/index.php?id=74