Спинальный дисрафизм (расщепление дужек позвоноков)
Закрытое расщепление дужек позвонков (spina bifida occulta) – врожденное отсутствие остистого отростка и различной части дужки. Отсутствие контакта мозговых оболочек или нервной ткани с внешней поверхностью
Два других состояния объединяют под названием открытое расщепление дужек позвонков (spina bifida aperta) или кистозное расщепление дужек позвонков (spina bifida cystica).
Менингоцеле – врожденный дефект дужек позвонков с кистозным расширением оболочек, но без аномалии нервной ткани. В 1/3 случаев имеется некоторый неврологический дефицит
Миеломенингоцеле – врожденный дефект дужек позвонков с кистозным расширением мозговых оболочек и структурными или функциональными нарушениями СМ или конского хвоста.
Закрытое расщепление дужек позвонков
Встречается у ∼20-30% жителей Северной Америки. Часто является случайной находкой, обычно не имеющей клинического значения, когда она наблюдается изолированно. Однако, в некоторых случаях она может быть связана с диастематомиелией, фиксированным СМ, липомой или дермоидной опухолью.
При наличии симптоматики, вызванной одним из сопутствующих состояний, проявления соответствуют фиксированному СМ (нарушения походки, слабость ног и атрофия, нарушения мочеиспускания, деформации стопы и др. Дефект может определяться пальпаторно; над ним могут быть кожные изменения (см. Кожные признаки дисрафизма в табл. 6-16).
Миеломенингоцеле
Эпидемиология/генетика
Частота расщепления дужек позвонков с менингоцеле или миеломенингоцеле (ММЦ) составляет 1-2/1000 живых новорожденных (0,1-0,2%). Риск возрастает до 2-3% в тех случаях, когда у одного, родившегося до этого ребенка, было ММЦ и до 6-8%, если у двух. Также риск повышен в тех семьях, в которых у близких родственников (братьев или сестер) рождались дети с ММЦ, особенно по материнской линии. Частота может увеличиваться во время войн, голода и экономических потрясений, но потом она постепенно снижается. Передача осуществляется не в соответствии с менделевскими законами, и, вероятно, является многофакторной.
Гидроцефалия при миеломенингоцеле
ГЦФ развивается у 65-85% пациентов с ММЦ. 5-10% пациентов с ММЦ имеют явную ГЦФ при рождении. У более чем 80% пациентов с ММЦ, у которых возникнет ГЦФ, это произойдет до достижения ими возраста 6 мес. Большинство пациентов с ММЦ имеют сопутствующую мальформации Киари 2 типа. Закрытие дефекта ММЦ может перевести латентную ГЦФ в активную за счет ликвидации пути оттока для ЦСЖ.
Табл. 6-14. Находки при миеломенингоцеле разных уровней
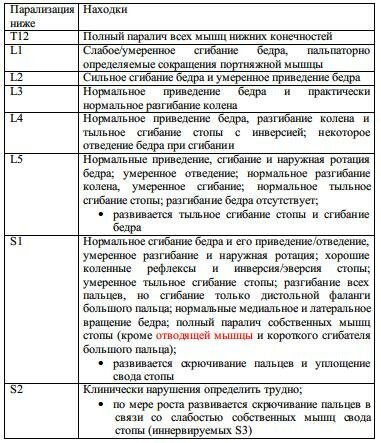
Исходы
Без всякого лечения могут выжить только 14-30% новорожденных; обычно это наименее пораженные пациенты. Из них 70% будут иметь нормальный индекс интеллектуального развития, 50% будут иметь возможность двигаться.
При современном лечении выживает ∼85% новорожденных с ММЦ. Наиболее частой причиной ранних смертельных исходов являются осложнения, связанные с мальформацией Киари (остановка дыхания, аспирация и т.д.). Поздние смертельные исходы обусловлены нарушениями функционирования шунтов. 80% пациентов имеют нормальные показатели индекса интеллектуального развития. Задержка умственного развития наиболее тесно связана с инфицированием, обусловленным шунтами. 40-85% пациентов могут перемещаться в корсетах, однако, большинство предпочитают передвигаться в кресле, т.к. это для них проще. 3-10% пациентов могут сами контролировать мочеиспускание, но в большинстве случаев для того, чтобы обеспечить сухое состояние кожи пациента, приходится производить периодические катетеризации.
Липомиелошизис
Дуральный спинальный дисрафизм с липомой. Описано 6 форм, следующие 3 клинически важны, как возможные причины прогрессирующих неврологических дисфункций в результате фиксации СМ и/или компрессии:
1. (интра)дуральная липома
2. липомиеломенингоцеле
3. фибролипома терминальной нити
Липомиеломенингоцеле
Подкожная липома, которая проходит через срединный дефект люмбо-сакральной фасции, дужку позвонка, ТМО и соединяется с ненормально низко фиксированным СМ. Соединение может быть терминальным, дорсальным или переходным (между предыдущими двумя формами).
Интрадуральная жировая опухоль также носит название липомы конского хвоста. В дополнение к тому, что конус СМ расположен ненормально низко, он расщеплен по средней линии по задней поверхности обычно на том же уровне, где имеет место расщепление дужек позвонков. Этот дорсальный миелошизис может продолжаться вверх под интактными дужками. Имеется толстая фибро-васкулярная связка, которая соединяется с наиболее ростральной расщепленной дужкой. Эта связка сдавливает мешок менингоцеле и нервную ткань, вызывая перегиб верхней поверхности менингоцеле.
На уровне дорсального миелошизиса ТМО имеет дефект, открывающий плакод. Липома проникает через этот дефект и прикрепляется к дорсальной поверхности плакода; она может распространяться вверх под нормальными дужками и может проникать в центральный канал выше уровня дорсального миелошизиса. Липома отличается от нормальной эпидуральной жировой клетчатки, которая является более рыхлой. САП обычно имеет выпячивание на стороне, противоположной липоме.
Клинические проявления
В серии наблюдений у детей в 56% случаев липома проявлялась в виде бокового объемного образования, в 32% случаев – нарушениями мочеиспускания, в 10% случаев – деформациями стопы, парализацией или болью в ноге.
Клинический осмотр
Практически все пациенты имеют кожные стигмы, которые сочетаются с расщеплением позвоночника: подкожные жировые комки (расположенные по средней линии и обычно распространяющиеся асимметрично в одну сторону) с/или без ямочек, родимые пятна, участки ненормального оволосения, открытый дермальный синус или кожные подвески. Может быть косолапость стопы.
50% больных могут иметь нормальный неврологический статус (большинство имеют только кожные проявления). Наиболее частым неврологическим нарушением является снижение чувствительности в сакральных дерматомах.
Диагностика
Обзорные спондилограммы пояснично-крестцового отдела в большинстве случаев показывают расщепление дужек (по определению должно быть практически во всех случаях, но в некоторых случаях вместо этого имеет место сегментирование). Также могут быть обнаружены аномальные соединения позвонков и дефекты крестца.
КТ/миелография или МРТ могут выявить ненормально низко расположенный конус. МРТ также показывает жировое объемное образование (высокий сигнал в режиме Т1, низкий в режиме Т2).
Всем пациентам перед операцией необходимо осуществить урологическое обследование для документирования возможных нарушений.
Дермальный синус
Тракт, начинающийся на кожной поверхности, выстланный эпителием. Обычно располагается на любом из концов нервной трубки: ростральном или каудальном. Наиболее частая локализация – люмбо-сакральная. Вероятно возникает в результате нарушения разделения кожной эктодермы с нервной эктодермой в период закрытия нервной бороздки.
Спинальный дермальный синус
Может проявляться в виде ямочки или синуса с/или без волос, обычно близко к средней линии, с внешним отверстием порядка 1-2 мм. Окружающая кожа может быть нормальной, пигментированной (цвета портвейна) или измененной в результате подлежащего образования.
Синус может заканчиваться поверхностно, может сообщаться с копчиком или может проходить между нормальными или расщепленными дужками позвонков к дуральному мешку. По своему ходу может иметь в любом месте расширение с образованием кисты. Киста называется эпидермоидной, если она выстлана слоистым ороговевающим эпителием и содержит только кератин от слущенного эпителия; либо дермоидной, если она выстлана дермой (имеет такие кожные придатки, как волосяные фолликулы и сальные железы) и содержит кожное сало и волосы.
Хотя внешне синус может быть мало заметен, он представляет угрозу проникновения инфекции в интрадуральное пространство с развитием менингита (иногда повторяющегося) и/или эндолюмбального абсцесса. В менее серьезных случаях может быть только поверхностная инфекция. Кожная выстилка содержит нормальные кожные придатки, которые могут приводить к тому, что внутри хода будут находиться волосы, кожное сало, слущенный эпителий и холестерол. В результате содержимое синусного хода становится раздражателем и может вызывать стерильный (химический) менингит с возможным отсроченным арахноидитом, если возникает сообщение с дуральным пространством.
Частота предполагаемого сакрального синуса (ямочка, дно которой не видно при разведении кожи): 1,2% от всех новорожденных.
Дермальный синус похож, но отличается от пилонидальной кисты, которая тоже может быть врожденной (хотя некоторые авторы считают ее приобретенной). Она содержит волосы, располагается поверхностнее постсакральной фасции и может инфицироваться.
Если ход продолжается интратекально и там формируется киста, то ее проявления могут быть похожи на фиксированный СМ или интрадуральную опухоль. Обычно первым проявлением в этом случае является нарушение функции мочевого пузыря.
Ход спинального дермального синуса по мере углубления всегда имеет направление в сторону головы. Окципитальный синус может проникать в череп и сообщаться с дермоидными кистами, расположенными в мозжечке или IV-ом желудочке.
Диагностика
Эти ходы нельзя зондировать или контрастировать, т.к. этим можно спровоцировать инфекцию или вызвать стерильный менингит.
Обследование направлено на выявление нарушений функций сфинктеров (анального и мочеиспускательного), люмбо-сакральных рефлексов, чувствительности и движений нижних конечностей.
Рентгенологическая диагностика
Если синус обнаружен при рождении, то лучшим способом определения расщепления дужек позвонков и наличия интрадурального объемного образования является УЗИ.
Если синус обнаружен уже после рождения, следует произвести МРТ. На сагиттальных изображениях можно увидеть ход и место его прикрепления. МРТ также является оптимальным методом, который показывает объемные образования (липомы, эпидермоиды и т.д.) внутри канала.
Узкие ходы, которые могут существовать между кожей и ТМО, нельзя увидеть на обзорных спондилограммах и КТ.
Обзорные спондилограммы показаны при подготовке к операции для возможности осуществления полной ламинэктомии.
Краниальный дермальный синус
Ход начинается с ямочки в затылочной или носовой области. Могут быть кожные признаки гемангиомы, подкожной дермоидной кисты, ненормального оволосения. Затылочный синус продолжается в каудальном направлении, и если он проникает в череп, то может достигать места стока венозных синусов. Проявлениями могут быть повторные бактериальные (обычно золотистый стафилококк) или асептические менингиты. Диагностика должна включать МРТ для выявления интракраниального распрстранения и сопутствующих аномалий, включая интракраниальные дермоидные кисты.
Гринберг. Нейрохирургия
Ю. А. Зозуля, Ю. А. Орлов
Институт нейрохирургии им. А. П. Ромоданова АМН Украины, г. Киев
Врожденные уродства развития являются одной из главных причин детской смертности и инвалидности. В Украине в 2001 году родилось почти 400 тысяч детей, из них 48 тысяч имели уродства. Значительное место в этой патологии занимают дефекты развития нервной трубки, которые формируют различные нарушения нервной системы: от пороков развития позвоночника и спинного мозга до анэнцефалии. При грубых дефектах развития невральной трубки (анэнцефалия, полное незаращение позвоночника и другие) плод погибает внутриутробно или рождается нежизнеспособным и погибает в ближайшие часы или дни после рождения. Поэтому социальный и медицинский аспекты грубых дефектов развития невральной трубки сводятся к профилактике формирования дефекта, его ранней диагностике и своевременному прерыванию беременности. Иные проблемы возникают при менее грубых нарушениях формирования спинного мозга и позвоночника, объединенных понятием спинальные дизрафии, или дефекты развития нервной трубки, которые в зарубежной литературе объединены термином spina bifida.
Историческая справка о спинальных дизрафиях
Исследования палеонтологов убедительно свидетельствуют о том, что врожденные пороки развития позвоночника и спинного мозга существуют также давно, как и человек. Известны описания дефектов развития позвоночника у взрослого человека неолитического периода (5000 лет до нашей эры), бронзового (3000 лет до нашей эры) и позднего железного века (800 лет до нашей эры).
Упоминания об опухолевых образованиях поясничной области мы находим в трудах Гиппократа (460–370 гг. до нашей эры). В работах итальянского анатома Морганьи Батиста (1688–1771 гг.) приведен обзор литературы XVI и XVII столетий, касающейся спинальных дизрафий, дано описание патологии дефектов невральной трубки с указанием связи spina bifida и гидроцефалии, spina bifida и анэнцефалии. Об этом пишут Pieter van Foreest (1522-1597 гг.), Nikolas Tulpii (1593–1674 гг.), Миколай Бидло (1714 г.). Лечению эта патология не подлежала, оно было бесперспективно.
XIX век открывает современную историю изучения спинальных дизрафий. В 1875 году R. Virchov убедительно доказал существование у человека скрытых незаращений позвоночника — spina bifida occulta. В 1881 году А. Лебедев на основании экспериментов на куриных эмбрионах и изучения человеческих плодов сделал вывод о том, что менингомиелоцеле и анэнцефалия являются крайними проявлениями одного и того же нарушения развития. Он также доказал возможность скрытых аномалий формирования нервной трубки. В 1886 году Recklinghausen опубликовал монографию, в которой подробно описал spina bifida как результат нарушения формирования нервной трубки, впервые выделил три ее вида: менингоцеле, менингомиелоцеле и миелоцистоцеле. Все работы исследователей носили описательный характер, хотя и связывали нарушения движений, недержание мочи, деформацию позвоночника и стоп с наличием дефекта развития нервной трубки — со spina bifida.
В доантисептический период лечение спинномозговых грыж сводилось к сдавливанию мешка и повторным проколам его. Рекомендованный Velpeau (1846) метод впрыскивания в полость мешка раствора йода не нашел распространения из-за частых осложнений и даже смерти пациентов. Более эффективный способ лечения был предложен доктором Вауer в 1889 году, который «закрывал» костный дефект выкроенным из подлежащих тканей мышечно-апоневротическим лоскутом. Предложенные в дальнейшем модификации этой методики остаются основными в хирургии спинномозговых грыж и в настоящее время. Однако до 50-х годов XX столетия отношение к хирургическому лечению спинальных дизрафий было отрицательным. В 1929 году J. Fraser опубликовал результаты хирургического лечения 131 ребенка в королевской детской больнице г. Эдинбурга (Англия). После операции выжили 82 ребенка. В течение года после операции еще 16 детей погибли от прогрессирующей гидроцефалии, большая часть выживших детей стали тяжелыми инвалидами. И вновь встал вопрос о целесообразности хирургического лечения спинальных дизрафий. Ситуация изменилась после внедрения в 50-х годах имплантируемых клапанных дренажных систем для лечения гидроцефалии (F. Nulsen, T. Spits, 1951; R. Pudenz, F. Russel, 1957). В сочетании с разработкой новых эффективных антибиотиков для лечения воспалительных осложнений, дренирующие операции, по существу, «открыли двери» для хирургического лечения спинномозговых грыж у детей, включая новорожденных. Однако это поставило новые проблемы перед ортопедами, урологами, неврологами, психологами. У детей часто обнаруживали парезы конечностей, деформации позвоночника и стоп, недержание мочи, задержку физического и психического развития, что требовало конкретного лечения. В 1957 году в Лондоне создано первое «Общество исследования гидроцефалии и spina bifida». По его примеру мультидисциплинарные группы медиков (нейрохирургов, ортопедов, урологов, неврологов, психиатров) для лечения детей со spina bifida были организованы во многих странах.
Значительный вклад в изучение хирургии уродств развития позвоночника и спинного мозга внесли российские ученые: А. А. Бобров (1892), П. И. Дьяконов (1893), В. И. Зиненко (1895), С. Губский (1902), В. Н. Парин (1913), А. Д. Сперанский (1925), Н. Н. Бурденко (1929), Б. Г. Егоров (1938), В. И. Ростоцкая (1954), Б. М. Рачков (1966), Г. В. Каргопольцева (1971), Н. Маджибов (1981), А. М. Шутов (1983), И. В. Зуев (1995), В. Г. Воронов (2000).
Много внимания уделяли вопросам диагностики и лечения детей со спинномозговыми грыжами украинские ученые: Ю. А. Свидлер (1962), Н. П. Гук (1965), Н. П. Масалитин (1978), Ю. А. Орлов (1993, 2000, 2003).
Классификация нарушений развития позвоночника и спинного мозга
Несмотря на то, что на связь наследственности и частоты спинномозговых грыж указывали еще исследователи XIX века, истинный интерес генетиков к этой проблеме появился в последние десятилетия XX века.
В настоящее время понятие «спинальные дизрафии» объединяет различные нарушения развития спинного мозга и позвоночника:
Скрытые незаращения позвоночника обычно локализуются в пояснично-крестцовой области и, как правило, клинически ничем не проявляются. Часто они являются случайной «находкой» при рентгенологическом исследовании позвоночника. Кожа в области незаращения дужки позвонка не изменена, но могут отмечаться пигментные пятна, подкожные жировики (липомы), свищевые ходы (дермальные синусы). Анатомическая сущность скрытой расщелины позвоночника состоит в неполном заращении дужки позвонка.
Со времени первых описаний скрытого незаращения позвоночника R. Virchow (1875), Recklinghausen (1886) считалось, что эта аномалия развития позвоночника, обусловленная нарушением окостенения, не требует медицинской помощи. По данным А. Д. Сперанского, опубликованным в 1925 году в работе «Происхождение spina bifida occulta в крестцовом отделе позвоночного столба человека», утверждалось, что неполное смыкание крестцовых дужек встречается у 70% людей и является нормой. Лишь последующие анатомические исследования и данные современных методов диагностики (компьютерная томография, ядерно-магнитная томография) позволили обнаружить сопутствующие изменения в местах дефекта дужек позвонков, которые приводят к ночному недержанию мочи, к болям в пояснично-крестцовой области, нарушению осанки, реже к слабости мышц ног, деформации стоп, чувствительным и трофическим нарушениям. Именно эти случаи spina bifida occulta требуют хирургической помощи.
Открытые кистозные расщепления позвоночника (истинные спинномозговые грыжи) в зависимости от степени вовлечения в патологический процесс нервных структур разделяют на следующие.
Содержимое грыжевого мешка — мозговые оболочки и ликвор (спинномозговая жидкость), форма его — обычно стебельчатая с суженной ножкой. Костный дефект захватывает обычно два-три позвонка. Каких-либо клинических проявлений при данной форме спинномозговых грыж не отмечается и только угроза разрыва грыжевого мешка, увеличивающиеся его размеры служат основанием для хирургической пластики дефекта.
Незаращение позвоночника и мягких тканей с несформировавшимся спинным мозгом (rhachischiasis posterior) является крайней степенью уродства, никогда не сопровождается кистозным компонентом и выпячиванием образования над кожей. Дефект кожи, мягких тканей, заднего полукольца позвоночного канала зияет, и в его глубине видна полоска нервной ткани с большим количеством мелких сосудов (area medullo-vasculosa). Дефект кожи прикрыт фрагментированной пиальной оболочкой с истечением ликвора. Частичный рахишизис у живых новорожденных обычно распространяется на 3-5 позвонков.
Типичным для всех видов и форм спинальных дизрафий является их заднее расположение с дефектом заднего полукольца позвоночного канала. Крайне редко (менее 1% случаев) незаращение формируется на переднебоковой поверхности канала, и возникают передние спинномозговые грыжи. При пояснично-крестцовой локализации эти грыжи распространяются в малый таз и затрудняют процесс дефекации. При более высоком расположении они могут сдавливать образования грудной клетки, шеи, носоглотки.
Расположение спинномозговых грыж по длиннику позвоночного столба в 90% случаев ограничивается пояснично-крестцовой областью. Грудная и шейная локализации грыж относительно редки. Интересно, что при исследовании материала спонтанных абортов японские ученые обнаружили более частое нарушение формирования позвоночника и спинного мозга в грудном и шейном отделах, а также высокую частоту дефектов, захватывающих весь позвоночный столб. Это, в определенной степени, говорит о том, что эмбрион и плод с грубым дефектом формирования невральной трубки, как правило, погибают.
Для понимания сущности формирования пороков развития позвоночника и спинного мозга необходимо, хотя бы в общих чертах, представить процесс эмбриогенеза этих структур. На первой неделе беременности у зародыша происходит деление клеток с образованием зародышевых узелков. На второй неделе — формирование внезародышевых частей и образование осевых органов зародыша. На третьей неделе идет процесс образования первичной невральной трубки из наружного зародышевого листка, который проходит стадии первичной (3-4-я недели беременности) и вторичной (4-7-я недели беременности) нейруляции.
Именно на этих этапах эмбриогенеза возникают первичные нарушения нейруляции и формирование спинальных дизрафий. В стадии вторичной нейруляции могут появляться пороки развития пояснично-крестцового отдела позвоночника. Поэтому ранние периоды беременности, если это не связано с наследственными факторами, являются определяющими для формирования дефектов развития невральной трубки, и все современные методы предупреждения этой патологии распространяются на периоды до наступления беременности и ее первые недели.
Современная диагностика пороков развития нервной трубки
Несмотря на успехи в ранней диагностике дефектов развития нервной трубки, благодаря внедрению в практику биохимических методик (исследование содержания α-фетопротеина и ацетилхолинестеразы в сыворотке крови матери и околоплодных водах), методов интраскопии плода (ультразвуковой, ядерно-магнитный) основное значение в снижении частоты этой аномалии принадлежит предупредительным мероприятиям. Учитывая, что причины возникновения дефектов развития нервной трубки многофакторные и эти факторы известны, обоснованно формирование групп риска беременных, у которых вероятность рождения ребенка с дефектом наиболее высока. Поэтому во всем мире признано, что при планировании беременности родителям необходимо обследоваться у врача-генетика, а будущей матери у гинеколога, чтобы предпринять меры по профилактике уродств развития нервной трубки, отнести беременных к различным группам риска и с различной настороженностью контролировать течение беременности.
Какие же факторы способствуют появлению дефекта развития нервной трубки?
Во-первых, генетический дефект, унаследованный от одного из родителей.
Во-вторых, воздействие неблагоприятных факторов внешней среды, способствующих появлению мутаций в гене. Известно, что встречаемость дефектов развития нервной трубки колеблется от 1:500 до 1:2000 живых новорожденных в различных регионах мира и этнических группах населения, составляя в среднем 1:1000. Однако, если в семье родителей или ближайших родственников встречались случаи рождения детей с дефектами нервной трубки, то вероятность появления ребенка с дефектом возрастает до 2-5%. Это же относится к рождению второго ребенка, если первый родился с дефектом (риск составляет около 5%). Настораживающим моментом в этом плане также являются спонтанные аборты (выкидыши), преждевременные роды, младенческая смертность в семье и у родственников.
Поэтому генетическая предрасположенность к появлению ребенка с дефектом нервной трубки является основным показателем включения беременной в группу высокого риска.
К внешним факторам, способствующим появлению дефекта развития нервной трубки, относятся:
Обнаружение одного, а тем более, нескольких из этих факторов, является основанием для включения беременной в группу высокого риска рождения ребенка с дефектом развития нервной трубки.
Оптимальный алгоритм пренатального обследования для снижения частоты дефектов развития нервной трубки предполагает следующее.
В период планирования беременности — консультации врача-генетика, терапевта, акушера-гинеколога, при необходимости уролога. Выделение групп беременных с высоким и низким риском рождения ребенка с дефектом развития нервной трубки.
Пренатальная диагностика и объем обследования беременных отличаются в различных группах риска.
В группах низкого риска проводятся:
В группах высокого риска проводятся:
Подтверждение дефекта развития нервной трубки — обычно основание для прерывания беременности, но современные методы пренатальной диагностики не являются абсолютными. Они чаще диагностируют сам факт наличия дефекта, однако не всегда можно уточнить степень выраженности его. В то же время, степень вовлечения в патологический процесс нервных структур считается определяющей для прогноза. При менингоцеле и своевременной хирургической помощи ребенок полноценно развивается, а в будущем становится нормальным трудоспособным человеком. При менингомиелоцеле даже хирургическая помощь не обеспечивает высокого качества жизни, ребенок будет инвалидом, нередко тяжелым. Поэтому обнаружение у плода дефекта развития нервной трубки — всегда веское основание для прерывания беременности.
Значительно сложнее ситуация в семьях, где беременность долгожданна, а перспектива новой беременности маловероятна. Если выраженность дефекта уточнить не удается, используют дополнительные методы диагностики: ядерно-магнитную резонансную томографию (МРТ), но и она не всегда позволяет ответить на поставленные вопросы. Тогда врачи совместно с родителями, объясняя все обстоятельства и возможные исходы, решают судьбу плода.
Статья опубликована на сайтеhttp://www.medolina.ru
Спинальный дизрафизм
http://radiopaedia.org/articles/spinal-dysraphism
Врожденные пороки позвоночника и спинного мозга
http://www.ajronline.org/doi/full/10.2214/AJR.07.7141
http://emedicine.medscape.com/article/413899-overview#showall
Спинальный дизрафизм
http://www.pediatricneurosciences.com/article.asp?issn=1817-1745;year=2011;volume=6;issue=3;spage=31;epage=40;aulast=Venkataramana