(Новости лучевой диагностики 1998 4: 13-15)
К вопросу диагностики гистиоцитоза из клеток Лангергарса.
Лазюк И. И.1, Борисевич Г. А.1, Сергеева А. А.1, Ратнер Т. П.2
1Кафедра лучевой диагностики в педиатрии БелГИУВ, 2Республиканский детский онкогематологический центр.
Заболевания, которые ранее называли по имени авторов, впервые описавших их (болезнь Гоше, Ниммана-Пика, Таратынова, Хенда-Шюллера-Крисчена, Леттерера-Сиве) в последующем были разделены на две группы:
1. Болезни накопления, в основе которых лежит наследственный дефицит ферментов, которые обеспечивают утилизацию липидов (болезнь Гоше, Ниммана-Пика);
2. Гистиоцитозы-Х (болезнь Таратынова, Хенда-Шюллера-Крисчена, Леттерера-Сиве), патофизиологическим субстратом которых является локализованная или диссеминированная пролиферация макрофагов без признаков атипии.
Исследования последних лет показали, что макрофаги из очагов поражения при гистиоцитозе-Х имеют структурные и функциональные характеристики клеток Лангергарса. В связи с этим международное общество гистиоцитологов в 1986 году предложило заменить термин гистиоцитоз-Х новым термином — гистиоцитоз из клеток Лангергарса (ГКЛ).
Этиология ГКЛ остается неизвестной. Согласно современной точки зрения это заболевание имунной регуляции с аномальным имунным ответом, приводящим к пролиферации клеток Лангергарса, образованию гранулем, эозинофильной инфильтрации, остеолитических очагов, фиброза и т.д. Для ГКЛ характерно хроническое течение со сменой обострений и ремиссий, иногда спонтанных.
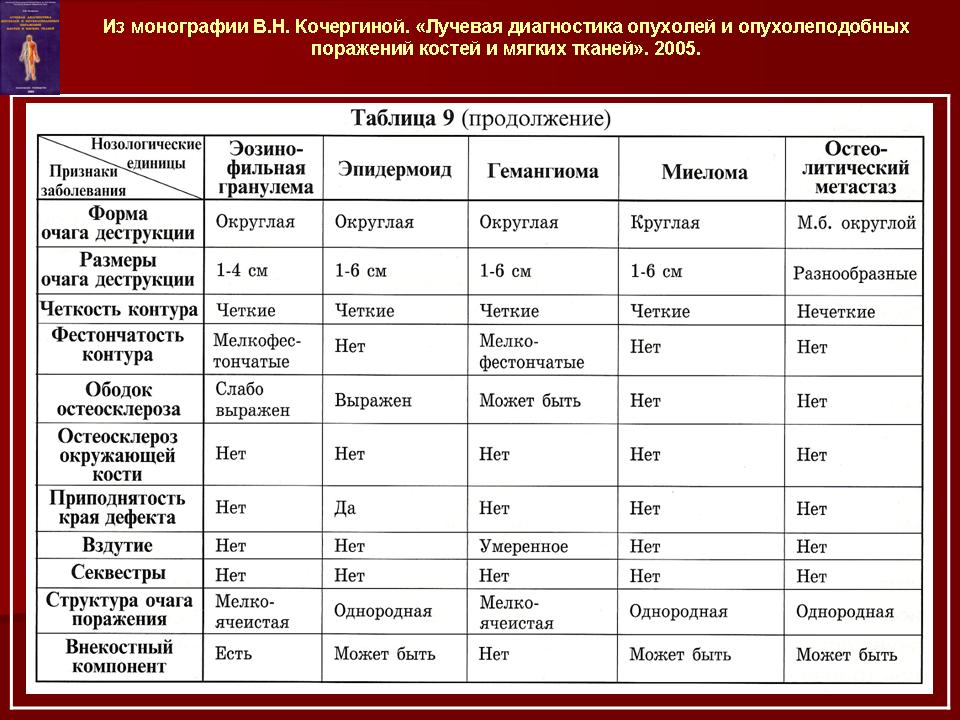
Эозинофильная гранулема или болезнь Таратынова характеризуется локальной деструкцией кости, обусловленной внутрикостным развитием гранулем, состоящих из гистиоцитарных клеток и эозинофилов. Обычно выявляется солитарная гранулема кости, реже — несколько очагов, и очень редко — множественные поражения скелета. Наиболее часто эти изменения локализуются в плоских костях черепа и в бедренной кости. Могут поражаться плоские кости таза, ребра, позвонки, челюсти.
Болезнь Хенда-Шюллера-Крисчена или распространенный хронический гистиоцитоз. При нем образуются множественные гранулематозные разрастания не только в костях, но и в коже, лимфатических узлах и внутренних органах. В типичных случаях характерна тетрада симптомов с несахарным мочеизнурением, прогрессирующим пучеглазием, увеличением печени и селезенки, поражением костей. Костные изменения чаще множественные, сопровождаются болевым симптомом в зоне деструкций. Особенно типичны изменения костей черепа. Ретроорбитальные опухоли при этом вызывают экзофтальм. Повреждения челюстей могут выводить зубы из альвеол. Поражение височной кости и сосцевидного отростка сопровождается развитием среднего отита. Очень часты поражения костей таза, ребер, лопаток, грудины, длинных трубчатых костей.
Болезнь Леттерера-Сиве или распространенный острый гистиоцитоз. Заболевание обычно проявляется на первом году жизни или при рождении ребенка. Это наиболее тяжелая, злокачественная форма процесса, характеризующаяся острым началом, бурным течением и быстрым летальным исходом. К ранним симптомам болезни Леттерера-Сиве относится себорейный дерматит с легкой инфильтрацией и сопутствующей пурпурой на волосистых покровах головы, ладонях, туловище, аденопатия, симптомы интоксикации, повышение температуры, повышенная кровоточивость, анемия, воспаление среднего уха, некротические тонзиллиты, увеличение печени и селезенки. Характерны также поражения костей черепа, таза, ребер, позвонков, длинных трубчатых костей. Рентгенологическая картина этих изменений не отличается от болезни Хенда-Шюллера-Крисчена.
Как за рубежом, так и у нас было доказано, что болезнь Таратынова, Хенда-Шюллера-Крисчена и Леттерера-Сиве не самостоятельные заболевания, а отдельные стадии одного и того же процесса.
Мы изучили материал, касающийся 29 больных гистиоцитозом, которые находились на обследовании и лечении в республиканском онкогематологическом центре за последние 15 лет.
Диагноз ГКЛ устанавливался на основании комплексной оценки клинико-рентгенологической картины заболевания и морфологического изучения биоптатов пораженных органов. До аварии на Чернобыльской АС (за 6 лет) их число составило 5, за такой же период после — 19, что свидетельствует о явной тенденции к увеличению заболеваемости ГКЛ среди населения республики. По нозологическим формам больные распределялись следующим образом:
- болезнь Таратынова — 5 (20%);
- болезнь Хенда-Шюллера-Крисчена — 18 (63%);
- болезнь Леттерера-Сиве — 6 (20%).
Таким образом, больные с распространенными формами ГКЛ составили 83%, что свидетельствует о несвоевременности диагностики данного заболевания (По данным зарубежных авторов число больных с распространенным поражением не превышает 54%). Среди наблюдаемых нами больных преобладали мальчики (60%). Дети в возрасте 1-3 года составили 34%.
При изучении анамнестических данных установлено патологическое течение беременности у большинства матерей, дети которых страдали ГКЛ. Из перенесенных заболеваний у 3-х детей отмечался длительно текущий двухсторонний гнойный отит, не поддающийся консервативной терапии, в связи с чем им проводились неоднократные антротомии. Длительность заболевания до установления правильного диагноза у 37% больных составила от 1 года до 2 лет.
Среди предъявляемых жалоб наиболее частыми были плохой аппетит, слабость, потеря массы тела, повышение температуры, увеличение периферических лимфатических узлов, жажда с полиурией, боль в суставах, голенях, позвоночнике.
При объективном исследовании более чем у половины больных (15 чел.) была выявлена специфическая ссыпь, у 17 детей — припухлость в области черепа и конечностей, у 5 — экзофтальм, что позволило уже при первичном обследовании заподозрить гистиоцитоз.
Со стороны периферической крови чаще всего регистрировалась анемия, лимфоцитоз, ускорение СОЭ (от 20 до 40 мм/час). Из произведенных 15 стернальных пункций характерную для гистиоцитоза картину удалось выявить только в 2-х случаях, что свидетельствует о малой информативности данного метода при ГКЛ. Гистологическое же исследование биопсийного материала из участков поражения оказалось эффективным в 17 из 18 произведенных биопсий.
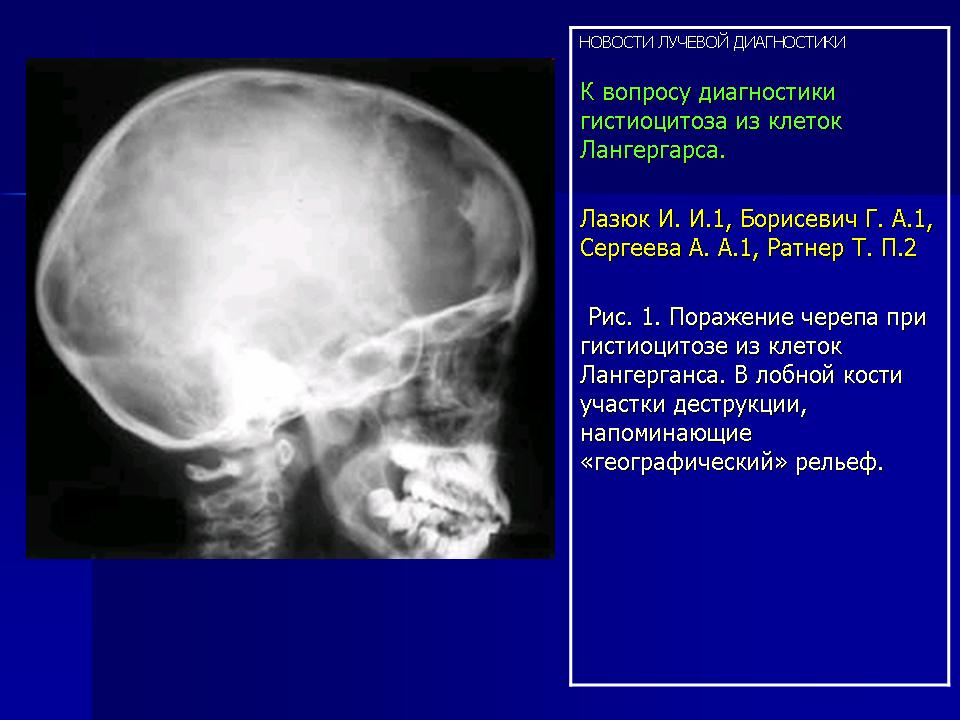
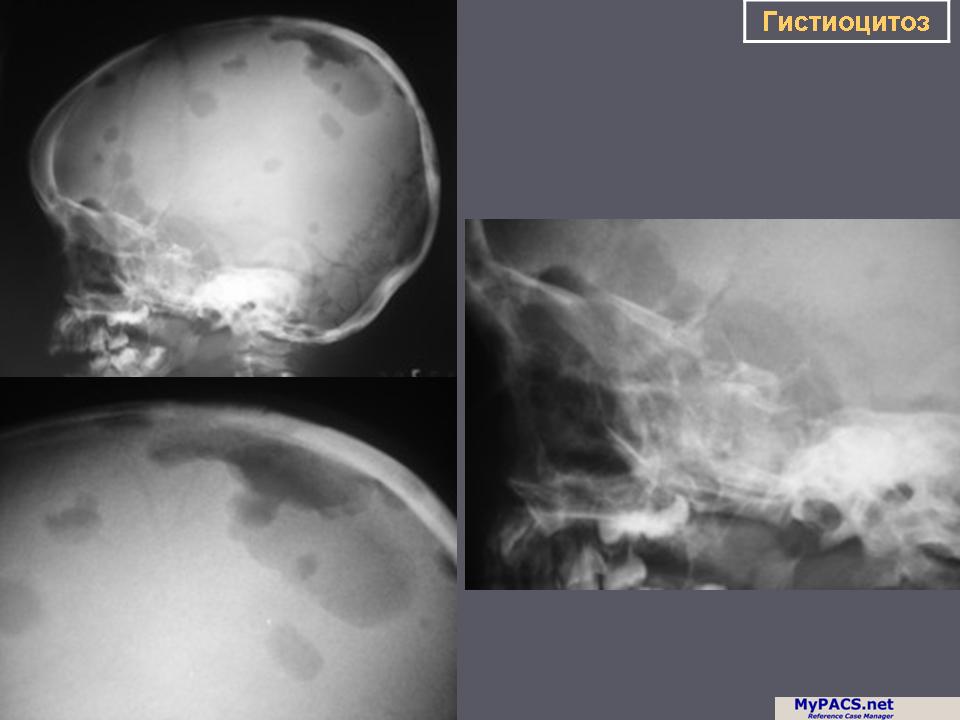
Изучение скелетограмм у 27 из 29 больных позволило выявить изменения, подтвердившие диагноз данного заболевания. Это указывает на высокую диагностическую ценность рентгенологического обследования при ГКЛ. Наиболее часто определялись поражения черепа в виде множественных (24) или единичных (3) дефектов в костях свода, где четко прослеживалась «штампованность» очагов. Дефекты не содержали секвестров, имели различную форму, величину, четкие наружные и неровные, фестончатые внутренние контуры. Периостальная реакция отсутствовала. При множественных участках деструкций картина напоминала «географический или ландкартообразный» череп, или «изъеденную молью ткань» (Рис.1).
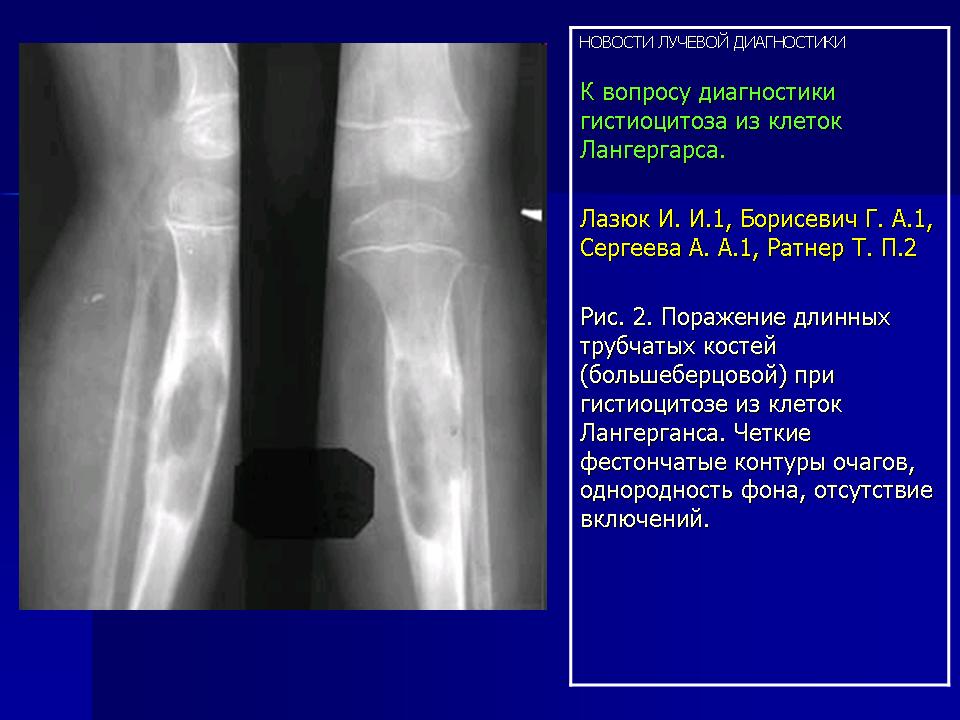
Наиболее часто изменения выявлялись в височных костях (21), несколько реже в лобных (16) и теменных (14). Реже всего поражалась затылочная область (4), орбита (3), сосцевидные отростки (2). В 44% случаев отмечались сочетанные поражения черепа и других костей, в том числе: ребер (17), подвздошных костей (6), ключиц (4), крыши вертлужной впадины (3), лонных и седалищных костей (3), бедренных (4) и большеберцовых (4). При поражении костей таза, ребер, ключицы определялись разновеликие дефекты, между которыми нередко сохранялись костные перемычки, создающие картину сетчатой или ячеистой структуры. В типичных проекциях гиперостоз улавливался не всегда или был прерывистым. Опорными рентгенологическими симптомами являлись четкие фестончатые контуры очагов, однородность фона, отсутствие включений. Поражения в длинных трубчатых костях (Рис. 2) чаще локализовались в метафизарных отделах проксимальных концов. Деструктивные участки обычно располагались субпериостально, имели овальную или округлую форму, четкие полициклические контуры. Наибольший их размер был направлен вдоль оси кости. Кортикальные слой изнутри истончался или прерывался. Снаружи определялись периостальные наслоения в виде скорлупы, имеющие иногда слоистый характер.
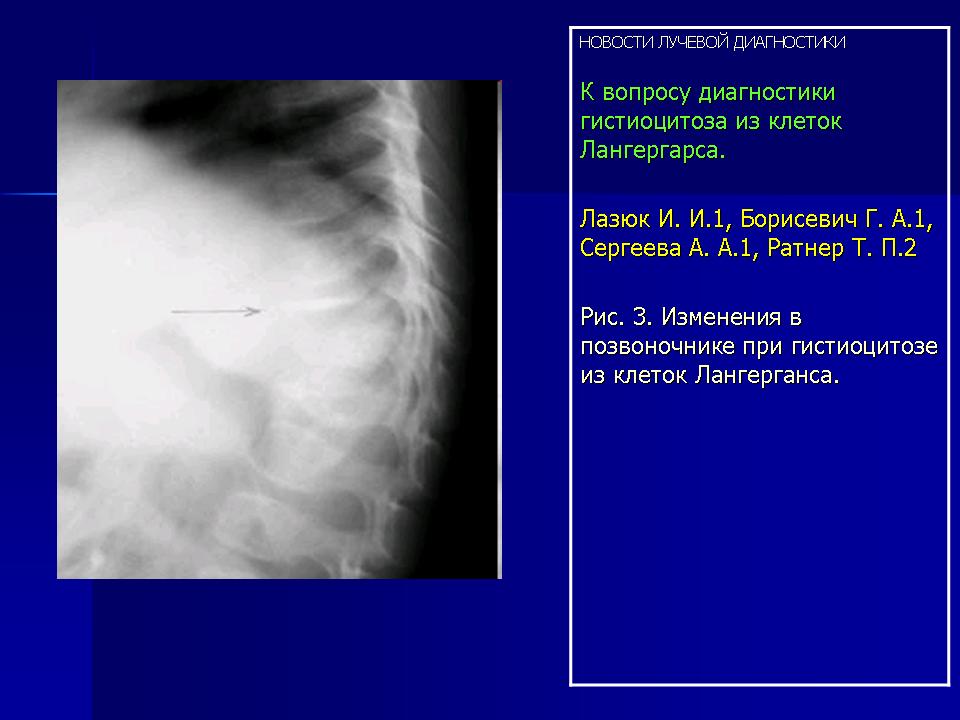
Изменения в позвоночнике наблюдались только в 2-х случаях. Тела позвонков при этом были сплющены до узкой пластинки, имели уплотненную структуру (Рис. 3).
Только у 2-х из 29 больных патология в костях отсутствовала. У одного из них выявлено генерализованное поражение внутренних органов, в другом случае диагностирована кожно-висцеральная форма ГКЛ.
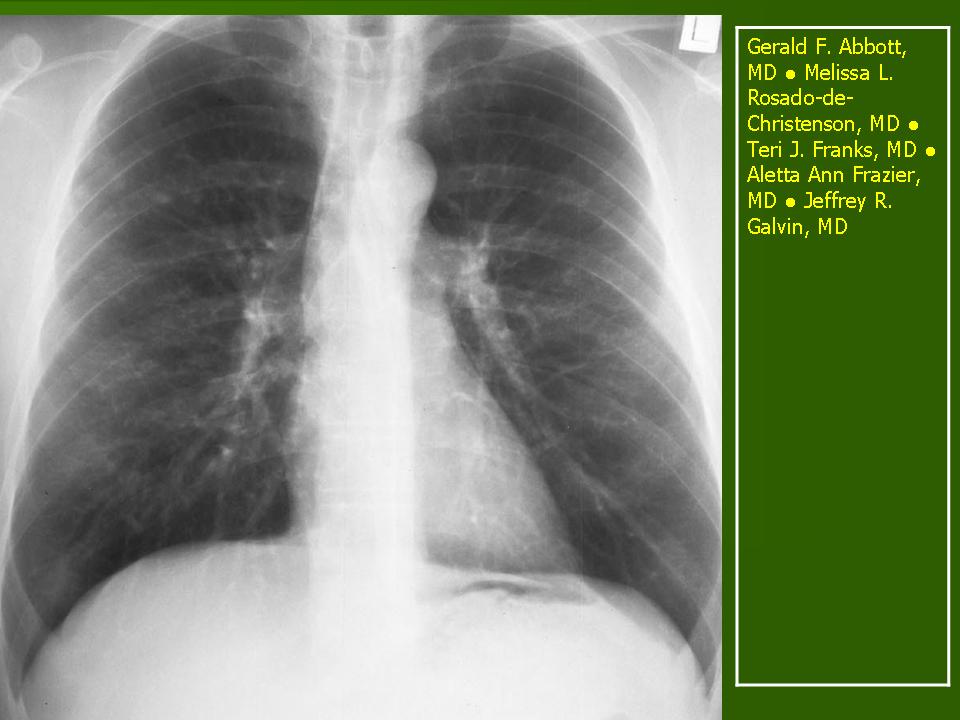
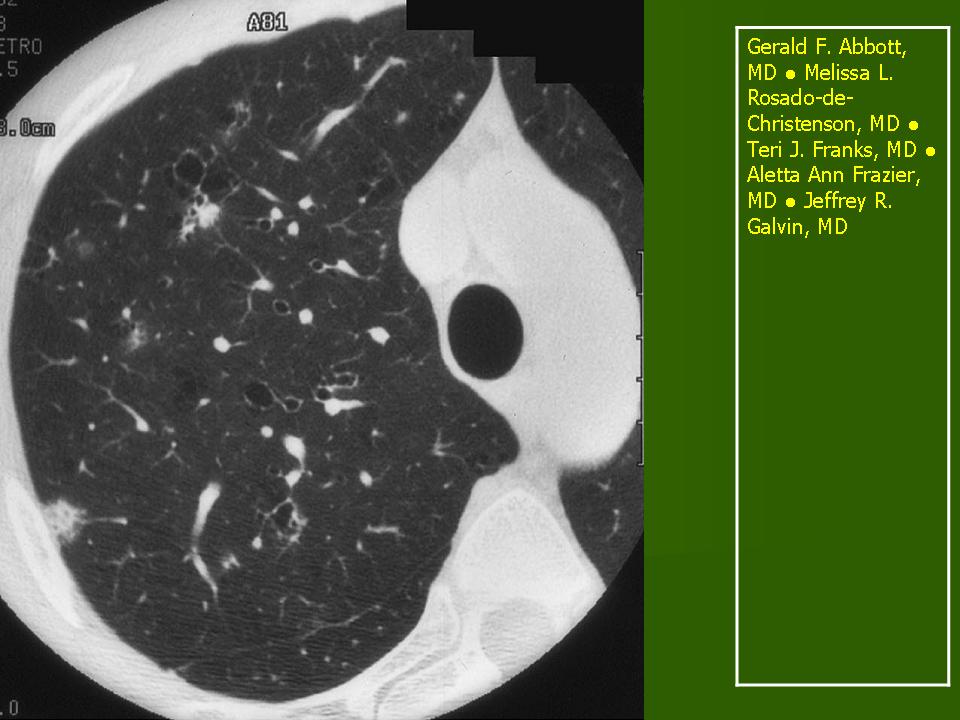
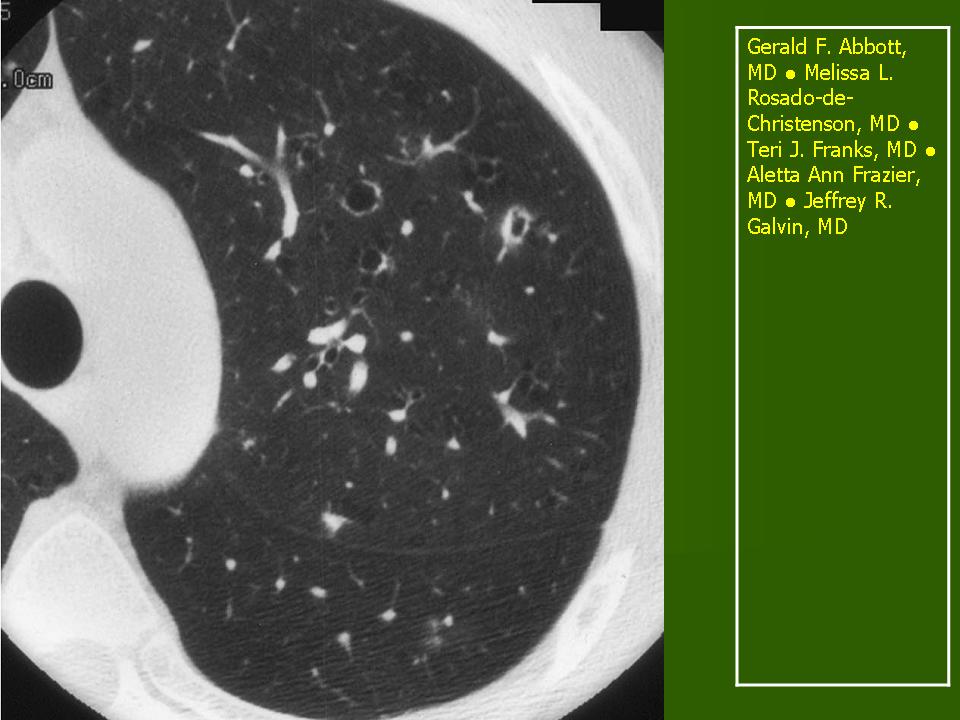
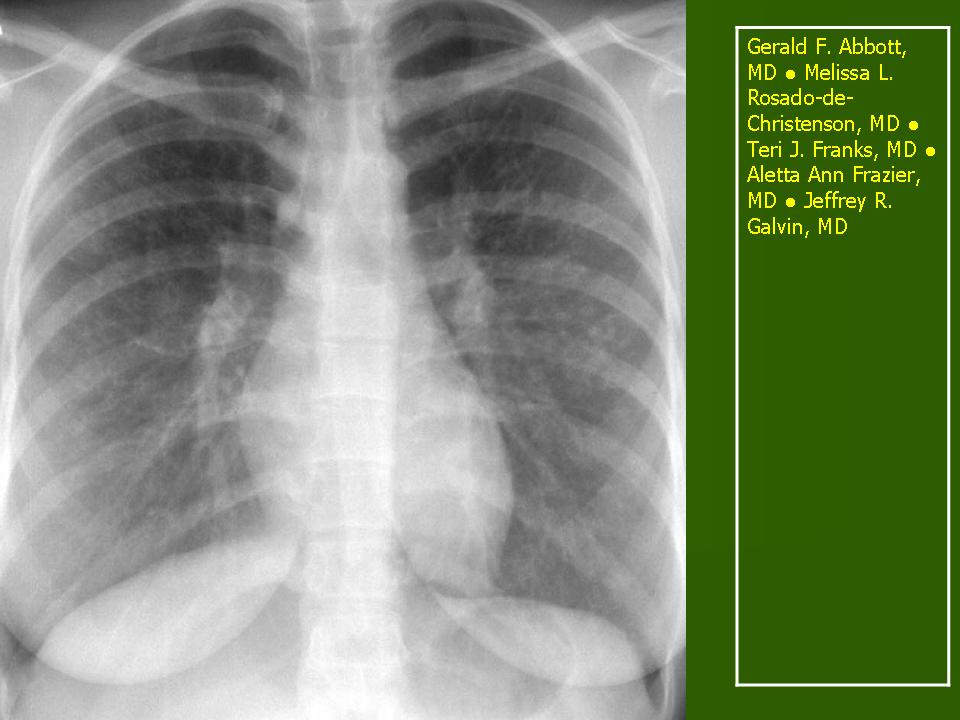
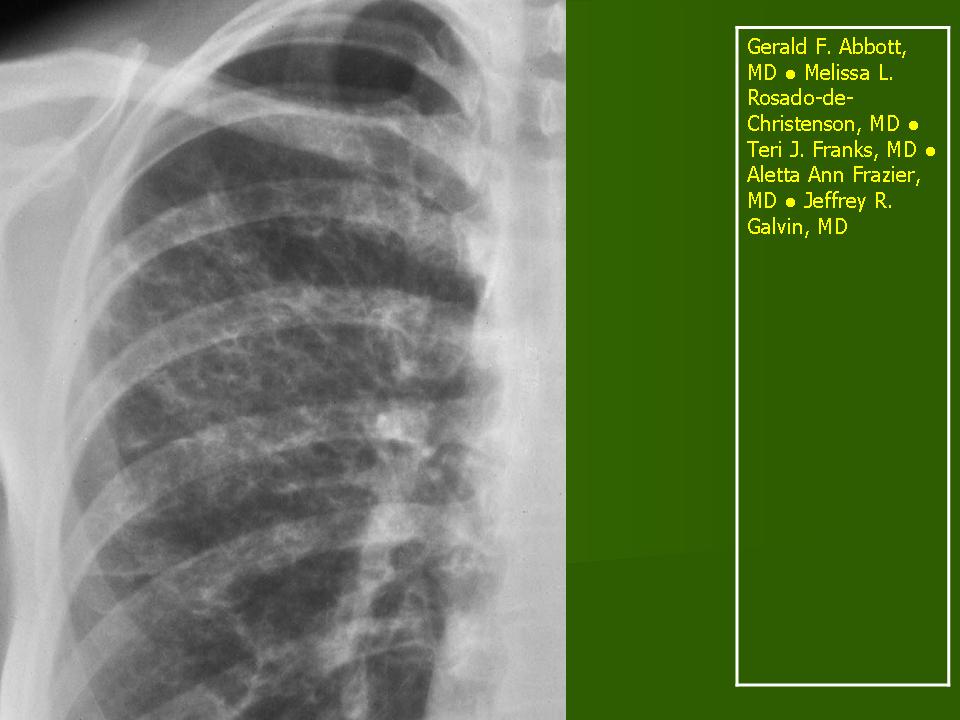
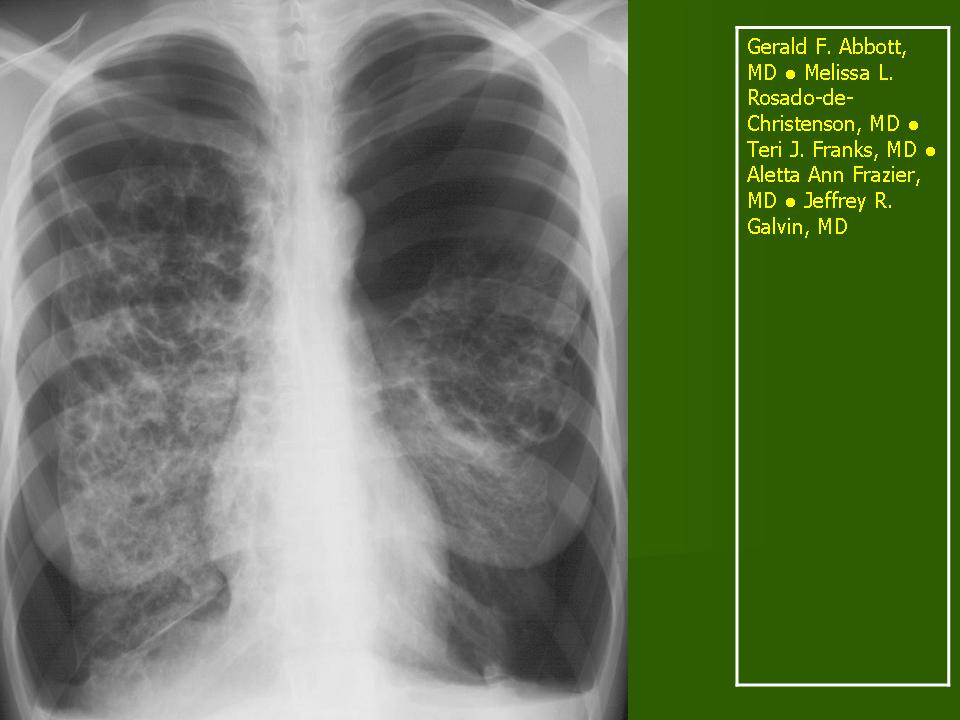
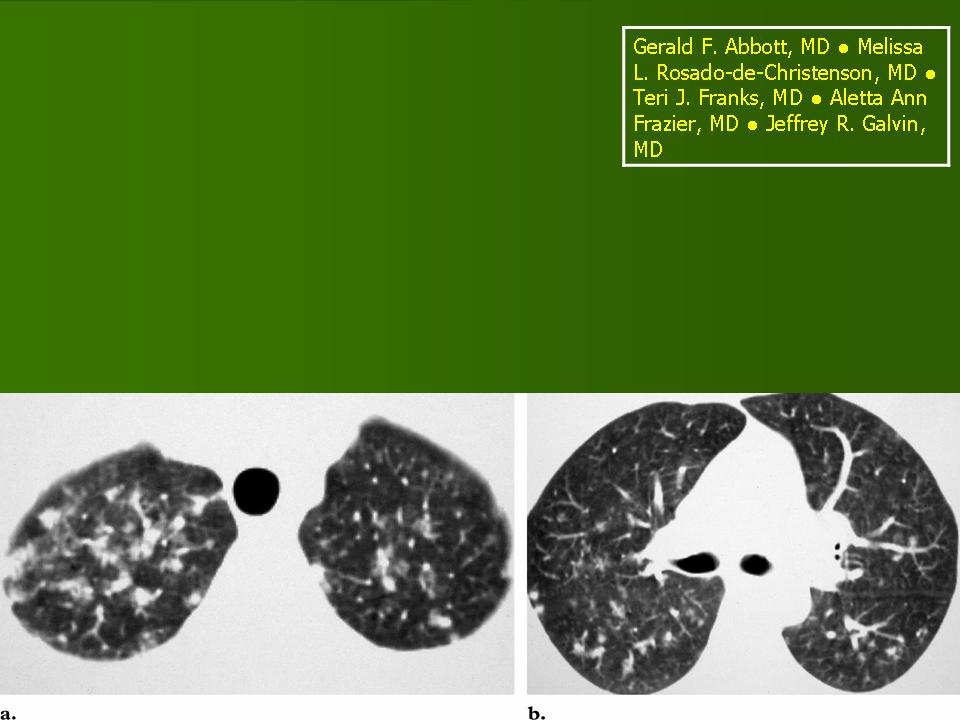
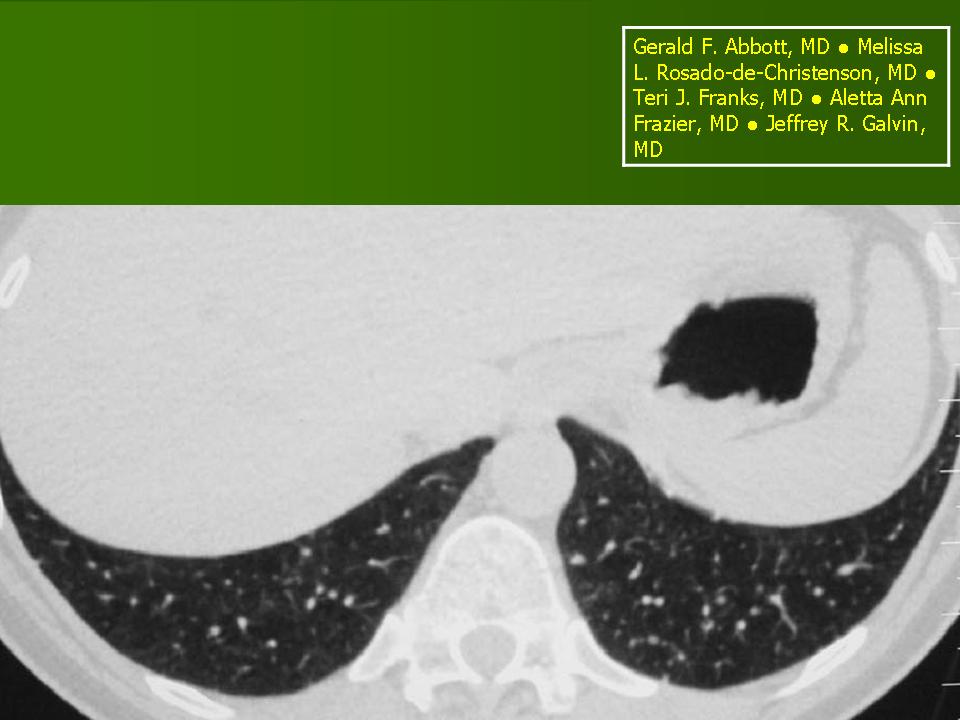
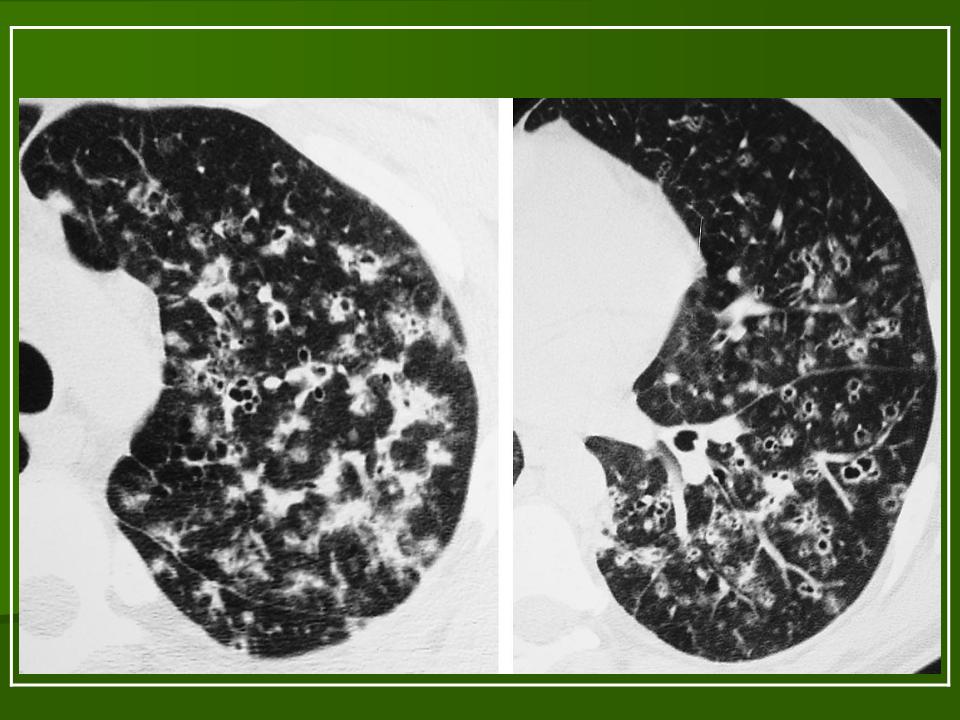
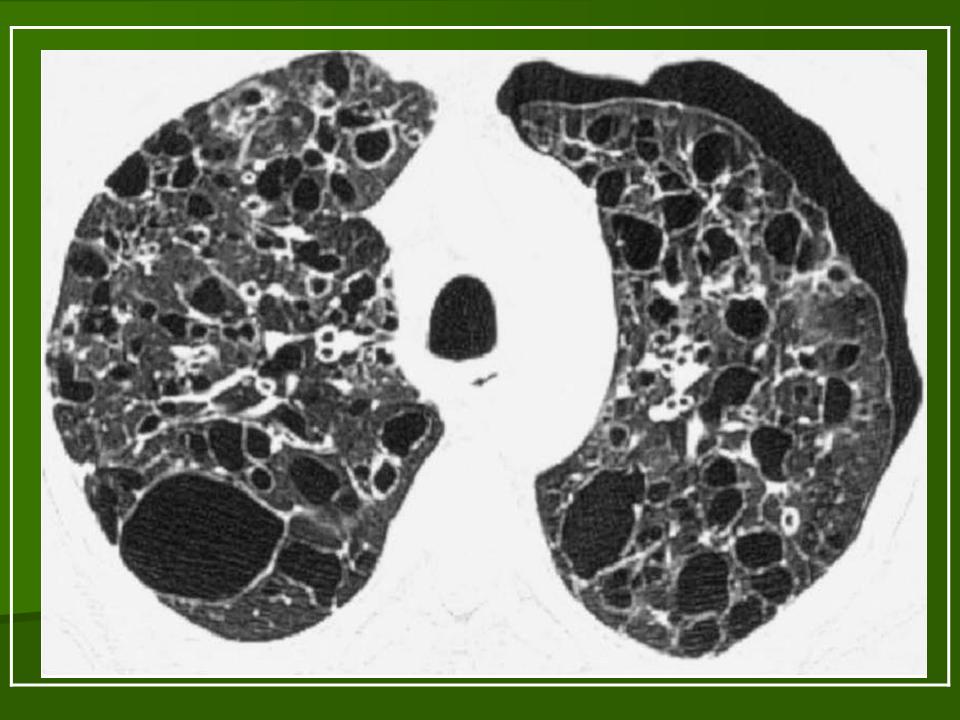
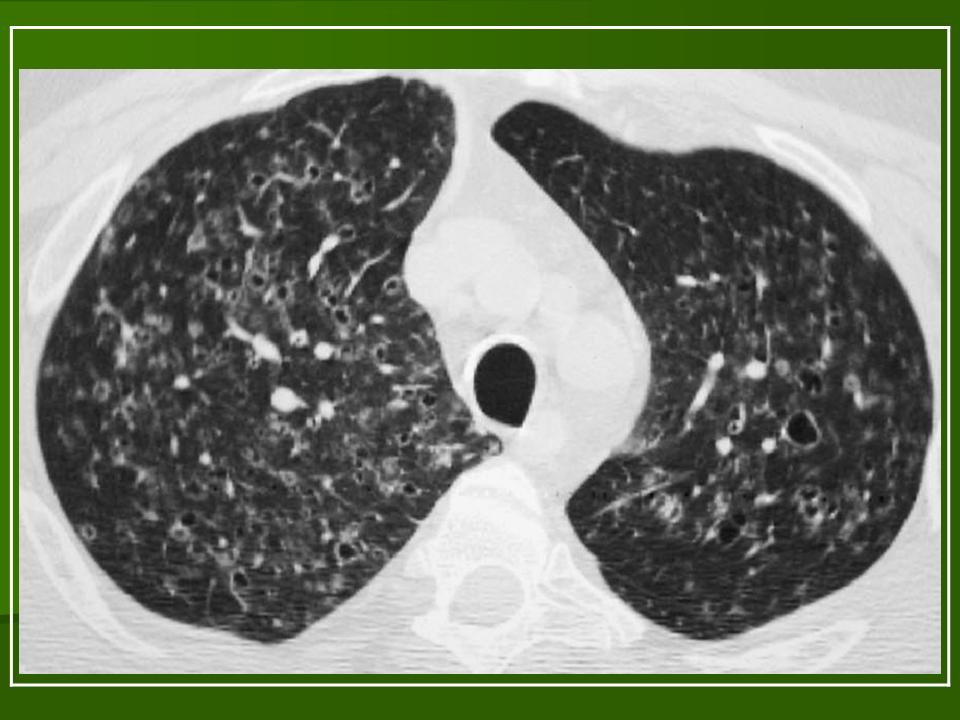
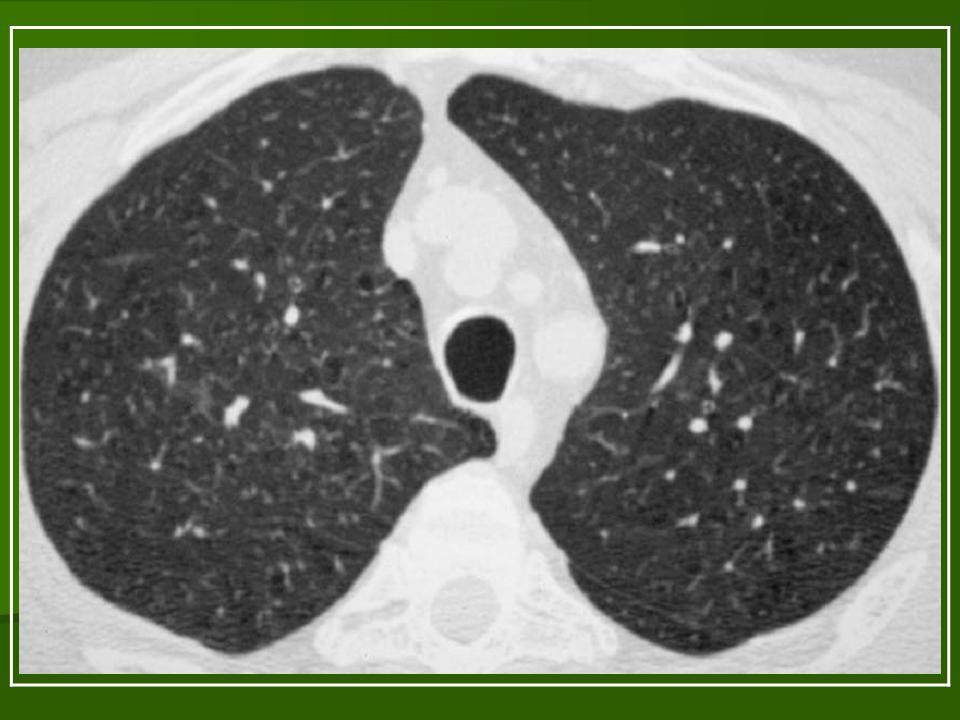
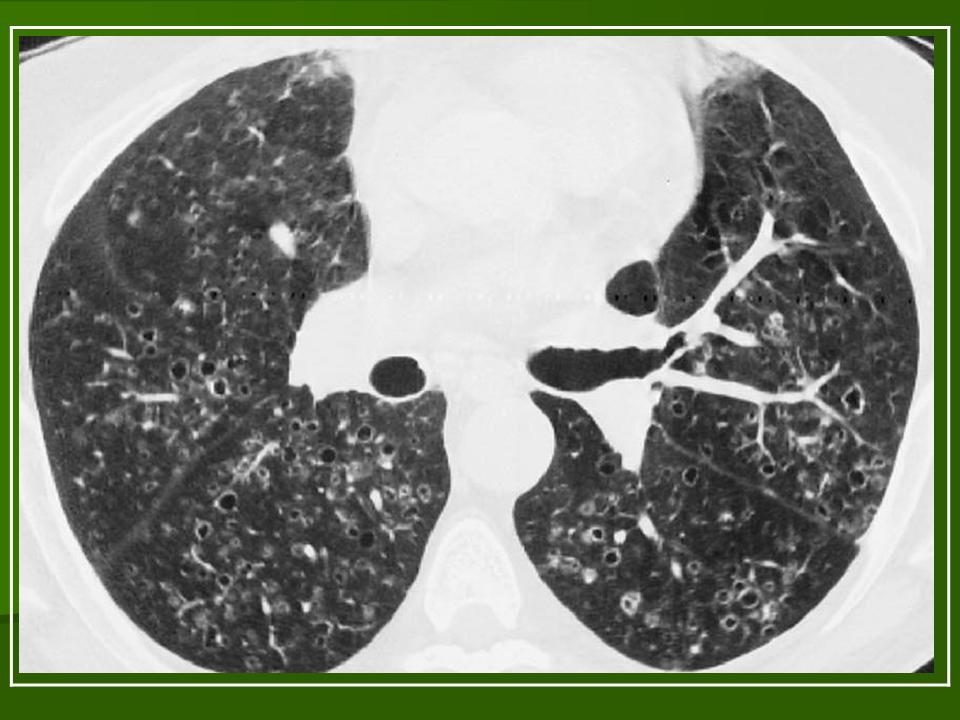
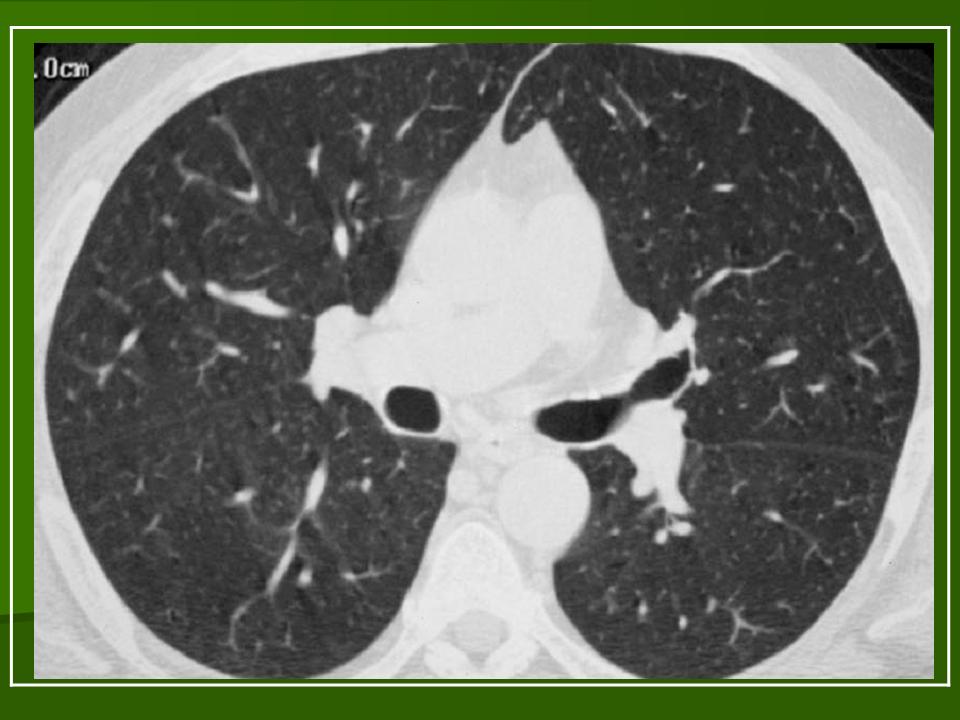
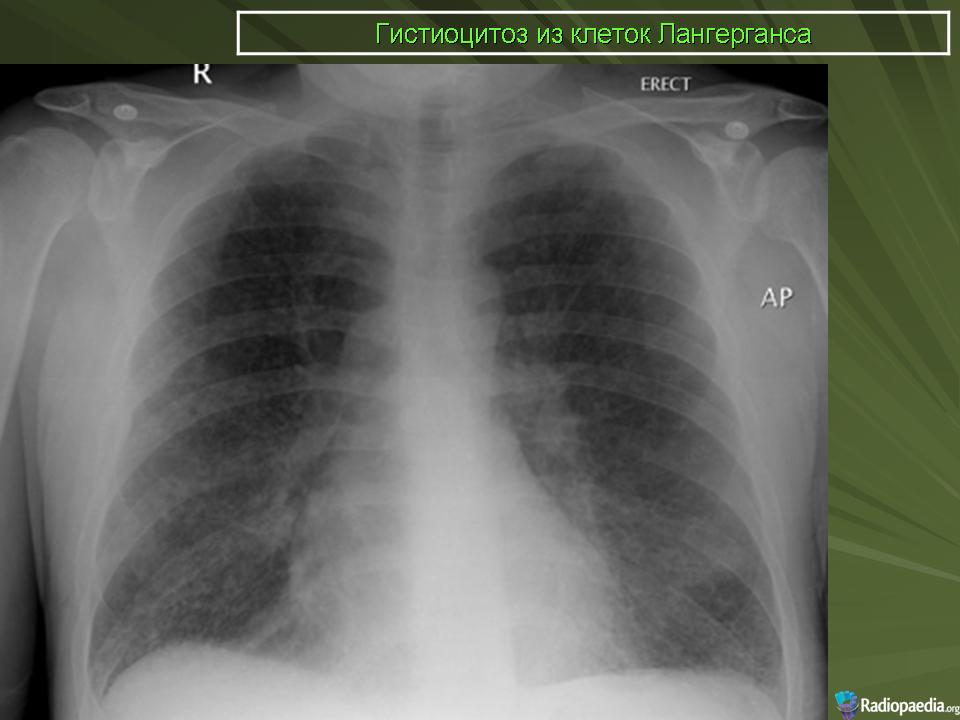
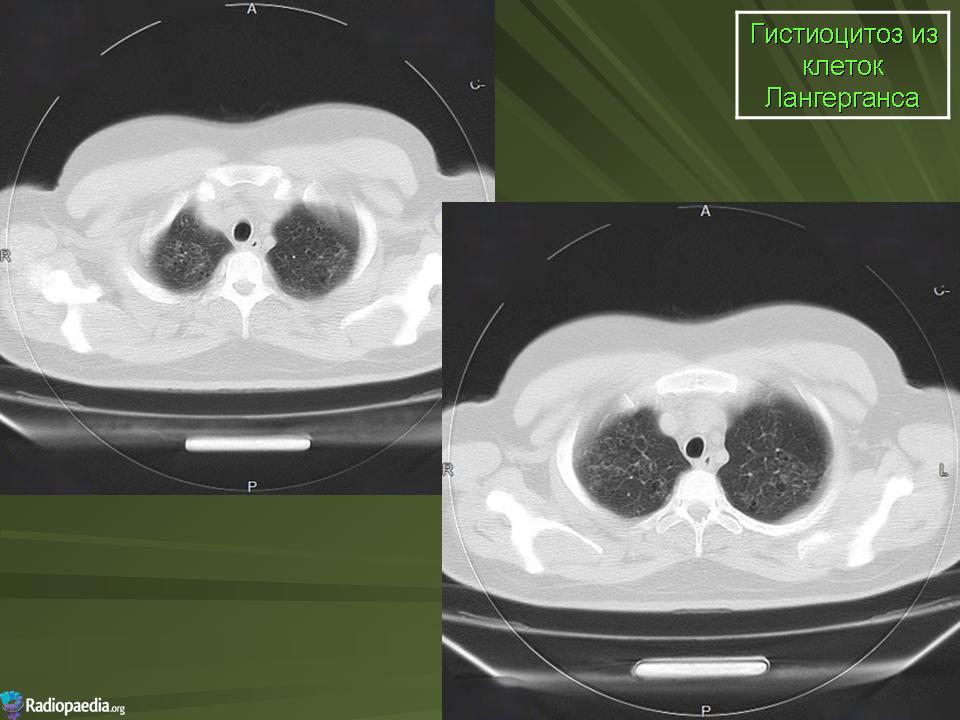
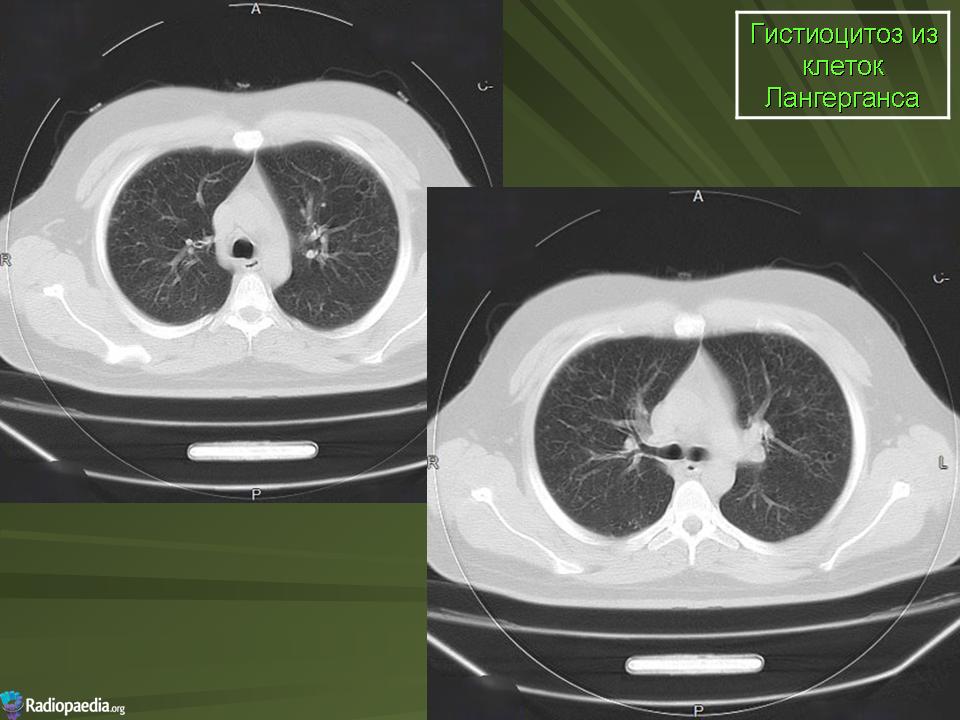
В 5 наблюдениях (21%) из больных с распространенной формой заболевания отмечались изменения в легких в виде обогащения, избыточности легочного рисунка, его деформации по сетчато-петлистому типу, очаговых теней. Корневая и медиастинальная аденопатия отсутствовали. В 1 случае в легких выявлена киста, которая периодически осложнялась нагноением (Рис. 4).
При лечении больных гистиоцитозом первоначально использовались схемы полихимиотерапии ЦОП или ЦОПП, в последние годы — усиленная программа ДАL-90. На фоне проводимой терапии 5 человек с установленной болезнью Леттерера-Сиве умерли, что составило 83% от больных с Леттерера-Сиве и 17% от всех больных с ГКЛ. При паталогоанатомическом вскрытии у 2-х из этих детей была выявлена дисплазия вилочковой железы, которая на наш взгляд явилась неблагоприятным фоном в течении заболевания. После проведенных терапевтических мероприятий положительная динамика с постепенной регрессией изменений зарегистрирована у 13 больных. У 10 человек (34%) были отмечены рецидивы заболевания в различные сроки (от 2 месяцев до 5 лет). Характерно, что при рецидивах процесса отмечалось поражение других участков скелета, вовлечение в процесс других органов и систем организма.
Таким образом, проведенное исследование позволило нам сделать следующие выводы:
1. Гистиоцитоз из клеток Лангергарса является редкой в республике патологией. Заболеваемость не превышает 0,2 на 100 тысяч населения.
2. Отмечается тенденция к увеличению заболеваемости после аварии на Чернобыльской АС.
3. Среди впервые выявляемых больных с ГКЛ преобладают лица с распространенными процессами, что свидетельствует о несвоевременной диагностике заболевания, обусловленной, на наш взгляд, недостаточным ознакомлением врачей общелечебной сети с данной нозологической формой.
4. При наличии у детей длительно текущих отитов, не поддающихся обычным методам консервативной терапии, следует исключить ГКЛ.
5. Рентгенологический метод исследования является одним из основных методов диагностики ГКЛ, позволяющий без использования дополнительных инвазивных методов высказаться в пользу данного заболевания.















Наблюдение Ola-la.
Наблюдение Nela.
Наблюдение Nela.
Наблюдение Ola-la.
Из литературных источников.
Продолжение.
Продолжение.
Продолжение.
Продолжение.
Продолжение.
ГИСТИОЦИТОЗЫ У ДЕТЕЙ
Гистиоцитозы представляют разнообразную группу заболеваний, для которых является характерным пролиферативный процесс в моноцитарно-макрофагальной системе. В зависимости от степени зрелости и дифференцировки гистиоцитарных элементов, существуют различные формы заболевания, имеющие особенности клиники, прогноза и лечения. Клеточные элементы системы мононуклеарных фагоцитов принимают участие в иммунном ответе против инфекционных и чужеродных антигенов с развитием гистиоцитарных пролиферативных синдромов реактивного или опухолевого характера.
Гистиоцитарные синдромы у детей
Класс I. Гистиоцитоз из клеток Лангерганса (гистиоцитоз X).
Класс II. Гистиоцитозы из мононуклеарных фагоцитов:
— семейный гемофагоцитарный лимфогистиоцитоз;
— инфекционно-ассоциированный гемофагоцитарный синдром;
— синусовый гистиоцитоз с массивной лимфаденопатией;
— болезнь (Rosai-Dorfman);
— ксантогранулема;
— опухоль-ассоциированный гистиоцитоз.
Класс III. Злокачественные гистиоцитарные заболевания:
— злокачественный гистиоцитоз;
— из «ординарных» фагоцитов;
— из интердигитирующих дендритических клеток;
— из клеток Лангерганса;
— истинно гистиоцитарная лимфома (гистиоцитарная саркома) из тех же клеток, что и злокачественный гистиоцитоз.
Гистиоциты (тканевые макрофаги) подразделяются на различные клеточные линии фагоцитарных и антигенпрезентативных клеток. К последним относятся интердигитирующие дендритические клетки, фолликулярные дендритические клетки и клетки Лангерганса. Выраженная пролиферация активированными клетками Лангерганса различных органов и систем представлена заболеванием — Лангергансово-клеточным гистиоцитозом. Ранее Lihtenstein было предложено название данного заболевания как гистиоцитоз X, куда он отнес следующие синдромы: болезнь Абта — Литтерера — Зиве, синдром Хенда — Шюллера — Крисчена и эозинофильную гранулему (болезнь Таратынова), которые различаются клиническими симптомами и прогнозом, но имеют одни и те же характеристики гистиоцитарной клеточной пролиферации.
Клетки Лангерганса характеризуются эозинофильной цитоплазмой, дольчатым или бобовидным ядром с наличием мелкодисперсного хроматина и небольших ядрышек. Кроме этого, в гранулематозном комплексе представлены лимфоциты, фагоцитирующие гистиоциты, эозинофилы и крупные макрофаги. Подтверждает диагноз иммуногистохимическое исследование с экспрессией CDIa антигена /ОКТ6/ или ультраструктурное исследование по выявлению гранул Бирбека в дендритических клетках.
В этиологии Лангергансово-клеточного гистиоцитоза существует несколько гипотез. Одной из них признана патология иммунной регуляции в результате цитокинно-медиаторной активации, возможно вследствие вирусной (ретровирусы) инфекции. Не исключается и опухолевая трансформация в характеристике пролиферативного процесса.
Диагноз основывается на клинико-рентгенологических данных и морфологическом изучении пораженных органов. По прогнозу больные подразделяются на две большие группы: с локализованными поражениями одной системы органов и с генерализованным поражением органов и систем. Худший прогноз у больных с дисфункциями гемопоэза, печени, легких. Локализованные поражения часто представлены изолированным поражением костной системы. Другими возможными очагами поражения являются лимфатические узлы, селезенка, кожа и реже — центральная нервная система.
По степени прогноза различаются:
0 степень — локальное поражение одной кости;
1 степень — изолированное поражение не более одной системы (наиболее часто — костной);
2 степень — поражение более одной системы без органной дисфункции;
3 степень — наличие органных дисфункций. К органным дисфункциям относятся:
— со стороны костного мозга — инфильтрация гистиоцитами, тромбоцитопения менее 100 000/мкл;
— со стороны печени — билирубинемия;
— со стороны легких — дыхательная недостаточность;
— со стороны ЦНС — несахарный диабет, неврологическая симптоматика.
Кости чаще поражаются у детей старшего возраста. В зависимости от локализации поражения, наблюдаются симптомы «экзофтальма», «гнойного отита», «гипертрофического гингивита», патологические переломы в области деструкции, компрессия тел позвонков. При рентгенологическом исследовании выявляются литические очаги с отчетливыми контурами (рис. 20).
Рис. 20. Поражение позвоночника при Лангергансово-клеточном гистиоцитозе.
Поражение кожи (рис. 21) часто наблюдается у детей раннего возраста и проявляется папулезной сыпью белесоватого или красно-коричневого цвета, иногда с изъязвлениями или образованием корочек. Типичная локализация на голове, туловище, реже — на конечностях. Нередко вторичное инфицирование кожных элементов.
Для поражения лимфатических узлов характерна локализация в шейных или паховых областях. Поражение печени сопровождается ее увеличением, гипербилирубинемией, гипопротеинемией, редко — асцитом. Вовлечение в патологический процесс селезенки характеризуется ее увеличением и симптомами гиперспленизма. Поражение печени и селезенки считается плохим прогностическим фактором. Гепатоспленомегалия характерна для детей раннего возраста.
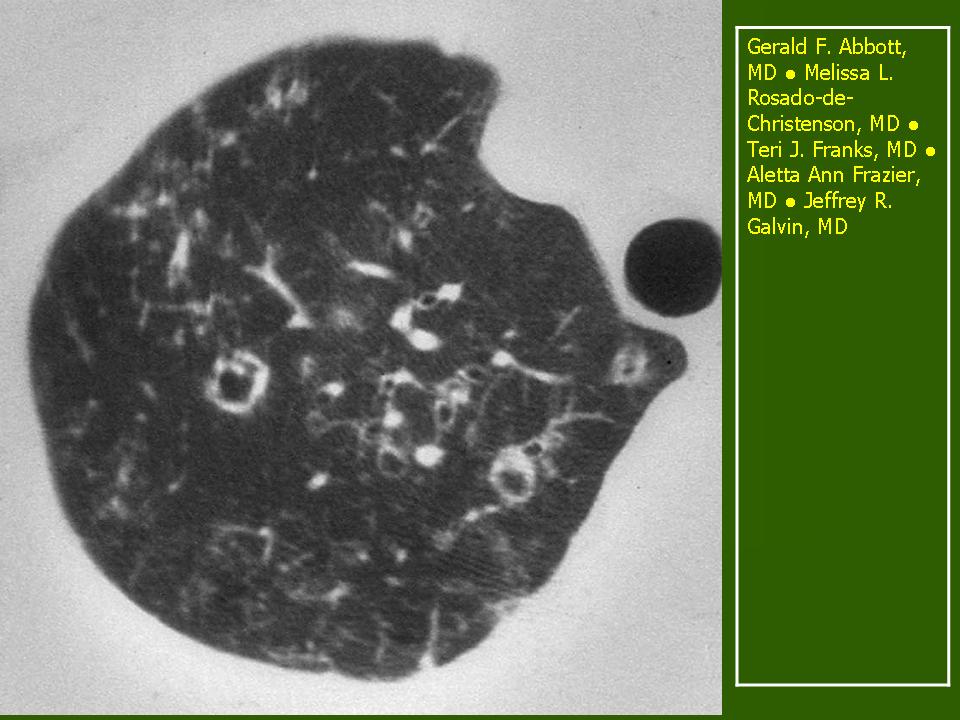
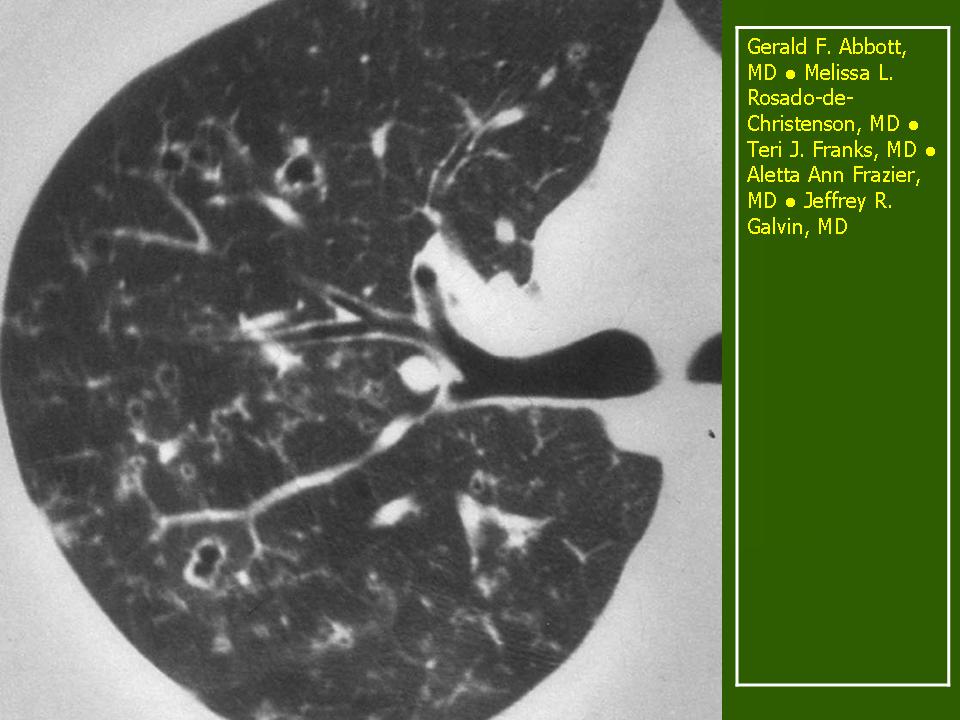
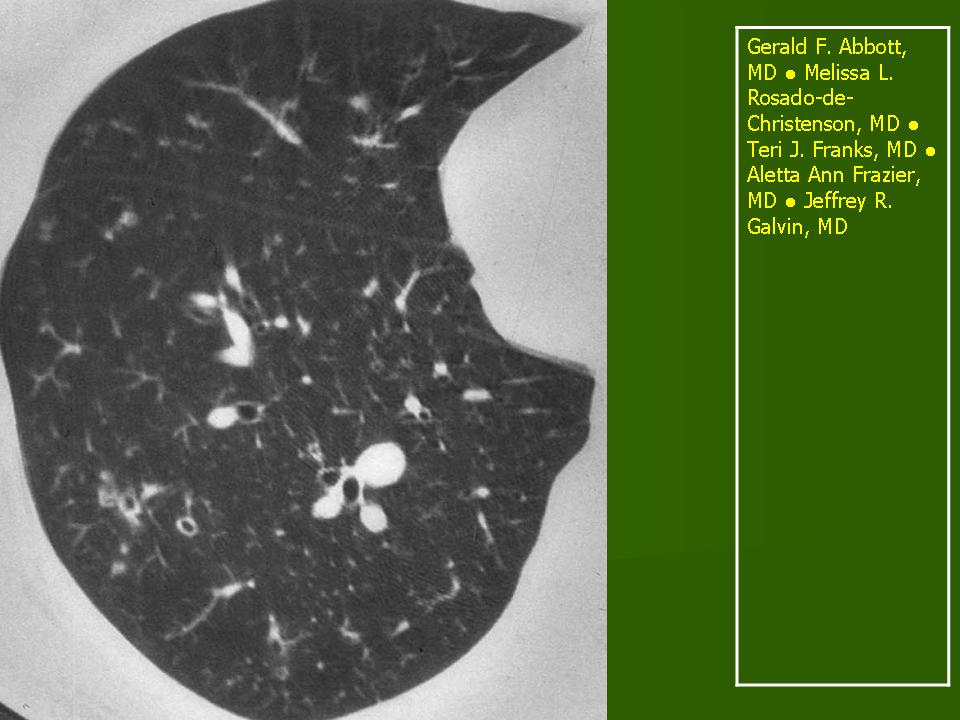
Поражение легочной ткани (рис. 22) свойственно любому возрасту. Часто больные имеют общие симптомы — лихорадку, слабость, реже — кашель, одышку, боли в грудной клетке. На рентгенограммах отмечается деформация и усиление легочного рисунка и микроузловые инфильтративные тени. Поражение костного мозга (с наличием клеток Лангерганса) сопровождается цитопенией периферической крови.
Рис. 21. Поражение кожи при Лангергансо-во-клеточном гистиоцитозе.
Рис. 22. Поражение легких при Лангер-гансово-клеточном гистиоцитозе.
Изменения в вилочковой железе морфологически характеризуются дистрофическими изменениями, вплоть до атрофии эпителия тимуса. Эти данные подтверждаются при секционном изучении тимусов. Желудочно-кишечный тракт вовлекается в патологический процесс редко и чаще у маленьких детей. Основными симптомами в данном случае являются энтеропатия, снижение питания.
Симптомы несахарного диабета и низкорослость (гипофизарный нанизм) возникают вследствие специфического поражения гипоталамо-гипофизарной области и обусловлены гипопитуитаризмом. Поражение центральной нервной системы встречается редко, в виде очаговых симптомов, диэнцефальных и церебеллярных нарушений.
Локализованные проявления заболевания отличает хороший прогноз. Заболевание у детей раннего возраста имеет, как правило, генерализованный характер с относительно плохим прогнозом.
Лечение Лангергансово-клеточного гистиоцитоза зависит от выраженности локализованных или генерализованных проявлений заболевания. Лечение генерализованных форм основывается на циклической полихимиотерапии. Ранее применялись различные сочетания глюкокортикоидов, винкалколоидов, алкилирующих агентов, антиметаболитов, эпиподофилотоксинов.Их применение способствовало выраженному положительному эффекту у 50—63% больных. В последнее время пристально изучается специфический для гис-тиоцитарных пролиферации препарат — этопозид (вепезид, VP-16). Однако использование последнего чревато отдаленными побочными эффектами. Наименее токсичным и достаточно эффективным препаратом является винбластин в стандартной дозировке и соответствующем режиме введения.
Программа химиотерапии гистиоцитоза из клеток Лангерганса (DAL-HX-83):
Индукция: преднизолон 40 мг/м2 с 1 по 40 дни;
вепезид 65 мг/м2 с 1 по 5 дни в/венно капельно;
винбластин 6 мг/м2 15, 22, 29 и 36 дни в/венно капельно;
вепезид 150 мг/м2 18, 25, 32, 39 дни в/венно капельно.
Поддерживающая терапия (54 недели):
1 степень— 6-меркаптопурин (6-МП) 60—80 мг/м2 ежедневно внутрь;
— преднизолон 40 мг/м2 1—5 дни внутрь на 9, 12, 15 и т. д. неделе;
— винбластин 6 мг/м2 в/венно капельно 1 день на 9, 12, 15 и т. д. неделе;
2 степень — то же лечение, что и при 1 степени с добавлением вепезида 150 мг/м2 в/венно капельно 1 раз на 5-м дне цикла 1 раз в 3 недели;
3 степень — лечение то же, что и при 2 степени, с добавлением метотрексата 500 мг/м2 в/венно капельно в течении 24 часов (с лейковорином) 1 раз в 3 недели на 1-м дне цикла.
Для быстрого купирования симптомов органных дисфункций изучается использование пульс-терапии повышенными дозами ме-тилпреднизолона в сочетании с винбластином. Изучается также использование иммуномодуляторов с применением альфа-интерферо-на и циклоспорина А. Дальнейшие наблюдения будут способствовать применению при данном заболевании более эффективных программ лечения.
Лангергансово-клеточный гистиоцитоз необходимо дифференцировать с другими заболеваниями, при которых наблюдается зрелоклеточная пролиферация гистиоцитов реактивного характера. Эти синдромы относятся ко II классу рабочей классификации гистиоцитозов у детей.
Синусовый гистиоцитоз с массивной лимфаденопатией впервые описан в 1978 году Rosai и Dorfman. В клинической картине заболевания преобладает увеличение шейных лимфатических узлов, реже — лимфоидной ткани носоглотки. Возможно поражение кожи, костей. Течение заболевания чаще непрерывно-рецидивирующее. Патогистологически проявляется пролиферацией макрофагов с признаками фагоцитоза плазматических клеток. Важным в патогенезе заболевания является нарушение иммунного ответа. Ультраструктурные исследования уточняют не-Лангергансово-клеточную природу гистиоцитов. Специфического лечения не требуется. Ранее с различными эффектами применялись химиотерапия (винкристин, циклофосфан, преднизолон) и лучевая терапия. В последнее время изучается применение препаратов интерферона (альфа- и реаферон) от 1 до 3 миллионов ME в/мышечно в течение 3—4 недель, с проведением повторных курсов. Прогноз заболевания благоприятный.
Вирус-ассоциированный гемофагоцитарный синдром является ответом организма на вирусную или другую инфекцию у больных с иммунодефицитным состоянием. Вирусы, приводящие к данному заболеванию, относятся к семейству герпесвирусов, аденовирусов, цитомегалии. У больных имеется пролиферация гистиоцитов с признаками эритрофагоцитоза в костном мозге, селезенке, печени и других органах. Клинические проявления в виде лихорадки, печеночной недостаточности, панцитопении, коагулопатии (ДВС-синдром), ухудшают прогноз заболевания.
Семейный гистиоцитоз с массивной лимфаденопатией является быстропрогрессирующим заболеванием, наследуемым по аутосомно-рецессивному типу, и характеризуется пролиферацией макрофагов. Болеют дети грудного возраста. Заболевание сопровождается лихорадкой, гепатоспленомегалией, панцитопенией. Поражаются: костный мозг, лимфатические узлы, селезенка, печень, мозговые оболочки. Отличается от вирус-ассоциированного гемафагоцитарного синдрома отсутствием связи с инфекцией и наличием наследственного фактора. Заболевание также фатально.
По современным данным, оба последних заболевания являются следствием нарушения продукции и контроля цитокинов, в частности, туморо-некротического фактора.
Во II класс гистиоцитозов относят и некоторые реактивные синдромы: болезни накопления (болезни Гоше, Ниманна-Пика, Фабри, ювенильная ксантогранулема и др.), большинство из которых являются наследственными липоидозами.
В III класс гистиоцитарных синдромов относят злокачественные опухолевые гистиоцитозы. Термин «злокачественный гистиоцитоз» был предложен в 1966 году Rappaport. По гистологической классификации злокачественных новообразований кроветворной и лимфоидной тканей ВОЗ, 1976 г. (Mathe, Rappaport) злокачественный гистиоцитоз отнесен к группе «острых лейкемий и родственных заболеваний», а гистиоцитоз из клеток Лангерганса (гистиоцитоз X) выделен в группу «хронический моноцитарный лейкемии и системных гистиоцитарных заболеваний».
Клинико-морфологическая характеристика злокачественного гистиоцитоза и гистиоцитоза из клеток Лангерганса различны. Различны также результаты лечения и прогноз заболеваний. Ряд авторов (Воробьев А. И. и др.) рассматривают злокачественные гистиоцитозы как макрофагальные опухоли и относят их к лейкозам, называя при этом макрофагальные лейкозы. Название данной опухоли пролиферации как гистиоцитарный вариант ретикулосаркомы или лимфомы и, тем более, одной из форм лейкоза едва ли является современным.
Термин «злокачественный гистиоцитоз» указывает на клинико-морфологическое отличие от гистиоцитоза из клеток Лангерганса. Диагноз устанавливается на основании морфологического исследования опухолевой ткани с помощью комплексной диагностики, включающей цитологический, иммуногистохимический, цитохимический, гистологический и субмикроскопический методы. Цитологическая характеристика опухолевых клеток представлена клеточным полиморфизмом. И. И. Матвеева выделила три типа опухолевых клеток: клетки с базофильной цитоплазмой и вакуолизацией, клетки вытянутой формы и эксцентричнорасположенным ядром, крупные клетки, содержащие фагоцитированные частицы с эритрофагоцитозом. Цитохимически клетки содержат высокую кислую фосфатазу, неспецифическую эстеразу, ингибируемую фтористым натрием. При ультраструктурном исследовании в опухолевых клетках отсутствуют гранулы Бирбека, отмечается выраженный фагоцитоз, в частности, эритрофагоцитоз. Пораженным клеткам присуще большое количество лизосом, фагосом, выраженный аппарат Гольджи, что отличает их от клеток Лангерганса
Клинически злокачественный гистиоцитоз характеризуется выраженными симптомами интоксикации в виде лихорадки и потери веса. Основные симптомы: лимфаденопатия, поражение костной системы, реже печени, селезенки, легких, плевры, кожи, почек, костного мозга, желудочно-кишечного тракта.
Первичной локализацией поражения у большинства больных являются лимфатические узлы шейно-надключичных, пахово-подвздошных областей, реже подмышечных, медиастинальных, брыжеечных. Отличительной особенностью поражения лимфатических узлов является инфильтрация мягких тканей с распадом и образованием изъязвлений на поверхности. Второй по частоте первичной локализацией поражения является костная система. При рентгенологическом исследовании выделяются мелкоочаговые деструктивные изменения в плоских и трубчатых костях, напоминающие метастазы опухоли; крупноочаговые с нечеткими контурами, сливные очаги деструкции и множественные очаги деструкции с четкими контурами, схожие с изменениями при миеломной болезни Реже первичной локализацией опухоли является кожа и подкожно-жировая клетчатка, мягкие ткани (например, орбиты, молочной железы, конечностей и пр.)
Лихорадка наблюдается практически у каждого больного и имеет разнообразный характер: от фебрильной до гектической, постоянный или периодический.
В клиническом течении заболевания у больных в короткие сроки наступает генерализация и диссеминация опухолевого роста с выраженностью симптомов интоксикации, похудания и поражением различных органов и систем. В периоде генерализации возможны изменения в легких в виде очаговой или множественной инфильтрации интерстиция. Для злокачественного гистиоцитоза более характерно поражение глубоких слоев кожи и подкожно-жировой клетчатки с образованием инфильтратов в виде узловатых образований синюшного цвета с распадом и изъязвлением в центре. Возможно выявление инфильтратов в молочных железах. Поражение костного мозга носит характер метастазирования по мере генерализации опухолевого роста, а не наоборот. В течение заболевания у больных нарастают проявления общей интоксикации в виде лихорадки с размахами температуры до 39—40'С, озноба, похудания. Особенности течения заболевания с дальнейшим метастазированием в различные органы дают право характеризовать заболевание как гистиоцитарную саркому.
Клинические стадии злокачественного гистиоцитоза (гистиоцитарной саркомы) укладываются в классификацию неходжкинских лимфом Murphy S., 1980 г. По данной классификации, больные, в основном, имеют III—IV клинические стадии.
При дифференциальной диагностике злокачественного гистиоцитоза от гистиоцитоза X, синусового гистиоцитоза с массивной лимфаденопатией, крупноклеточных лимфосарком (иммунобластной, макролимфобластной), а также низкодифференцированного рака, меланомы, рабдомиосаркомы и т. д., помимо микроскопических признаков, использовали детали ультраструктуры опухолевых клеток: отсутствие гранул Лангерганса, в значительно меньшей степени выраженный фагоцитоз позволяли исключить гистиоцитоз X. Высокая степень дифференцировки всех составляющих пролиферат клеток, фагоцитоз лимфоидных, плазматических клеток, сегментоядерных лейкоцитов, а не эритроцитов, были характерными чертами синусового гистиоцитоза с массивной лимфаденопатией. Отсутствие десмосом, пучков тонофиламентов позволяли отвергать недифференцированный рак, меланосом и промеланосом — меланому (метастаз), миофиламентов — рабдомиосаркому и т. д.
Особенности клинических проявлений, наличие в пролифератах различной степени зрелости гистиоцитарных клеток с преобладанием бластных форм дают право считать это гистиоцитарными опухолями с определенной морфологической остротой процесса, т. е. острым гистиоцитозом. Наличие у большинства больных в начале заболевания первичной локализации опухоли с дальнейшим в течение болезни метастазированием в различные органы и системы дают право характеризовать заболевание как гистициотарную саркому.
Е.Ф. Аргунова, А.П. Боброс, Е.С. Банщикова, О.Н. Иванова, М.В. Самаркина, С.А. Кондратьева
Представлены данные о 25 детях с диагнозом гистиоцитоз из клеток Лангерганса (ГКЛ) за период с 1998 по 2008 гг. Моносистемный ГКЛ диагностирован у 18 (72%), мультисистемный - у 7 (28%) детей. При моносистемном ГКЛ преобладает поражение костной системы. Полисистемный ГКЛ наблюдается у детей раннего возраста, тяжесть состояния и прогноз зависят от поражения «органов риска». Химиотерапия проведена 12 детям с моносистемной полифокальной и системной формами ГКЛ. Использовались международные протоколы лечения. При моносистемной форме ГКЛ прогноз благоприятный, при системном ГКЛ лечение оказалось не достаточно эффективным.
Введение.Термин «гистиоцитоз из клеток Лангерганса» (ГКЛ) введен в 1985 г., хотя болезнь была описана более ста лет назад, на первой встрече Международного общества по изучению гистиоцитозов для обозначения заболевания, которое ранее описывалось как болезнь Леттерера-Зиве, синдром Хендера-Шюллера-Крисчена, эозинофильная гранулема и гистиоцитоз Х. Данное заболевание является результатом пролиферации и диссеминации патологических гистиоцитов с признаками нормальных клеток Лангерганса [1, 3]. ГКЛ является достаточно редким заболеванием, ежегодная частота возникновения ГКЛ составляет от 0,5 до 2 случаев на 100 тыс. детского населения [2]. В 1987 г. была разработана рабочая классификация гистиоцитозов, уточнена терминология и критерии диагностики отдельных нозологических форм. Это дало возможность продвинуться в понимании данной относительно редкой патологии, в изучении ее клинических особенностей и разработке эффективных методов лечении [1, 2, 3]. В настоящее время общепринято разделение заболевания на моносистемное, поражение одной системы, и полисистемное, множественное поражение органов с нарушением их функций или без него. Это разделение отражает различный прогноз, следовательно, и различный подход к лечению и наблюдению. Прогностическое значение имеет поражение, по крайней мере, одного из «органов риска», а именно органной дисфункции печени, легких, костного мозга и/или селезенки [1]. Международные кооперированные исследования по лечению ГКЛ проводятся по унифицированным протоколам с использованием преднизолона, винбластина, вепезида: DAL-HX-83, DAL-HX-90, LCH-I, LCH-II, LCH-III. Для определения тактики ведения больных, не ответивших на стандартное лечение по протоколу, предложены комбинированная химиотерапия с включением цитозара и кладрибина, трансплантация стволовых клеток [1, 3].
Целью настоящей работы явилось изучение ГКЛ у детей РС (Я).
Материал и методы исследования: проведен ретроспективный анализ историй болезни 25 детей в возрасте от 1 года до 15 лет, находившихся в гематологическом отделении ПЦ РБ №1-НЦМ с диагнозом ГКЛ с 1998 по 2008 гг. Всем больным проводились лабораторные и инструментальные методы исследования, в т. ч. рентгенологические, компьютерная томография (КТ), магнитно-резонансная томография (МРТ). Проводились биопсии мягкотканого компонента, замещающего очаги деструкции костной ткани, кожи, лимфатических узлов, селезенки, с гистологическим исследованием. Химиотерапия проведена 12 детям с моносистемной полифокальной и системной формами ГКЛ. Использовались международные протоколы лечения.
Результаты и обсуждение. За истекший период выявлено 25 детей с диагнозом ГКЛ. Из них 11 детей младше 2-х летнего возраста. Выявляемость первичных больных от 1 до 4 случаев в год. Соотношение мальчики/ девочки составило 1,5:1,0. Сельских - 8 (32%) детей, городских - 17 (68%), в том числе из г. Якутска - 13 (52%). Моносистемный ГКЛ диагностирован у 18 (72%), мультисистемный - у 7 (28%) детей.
При моносистемном ГКЛ изолированное поражение лимфоузла диагностировано у 1-го ребенка, поражение костной ткани у 17 детей. Костная система поражалась в виде единичных или множественных очагов деструкции кости литического характера. Один очаг костной деструкции (монофокальное поражение) выявлен в 9, более 2-х очагов (полифокальное поражение) в 8 случаях. При полифокальном ГКЛ деструктивный процесс в 2-х костях установлен в 5 случаях, в 3-х и 6 костях по 1 случаю. По нашим данным чаще поражались кости черепа, теменная, лобная части (11 случаев) (рис.1) и трубчатые кости нижних конечностей (7) (рис. 3), реже челюсти, подвздошная кость, грудина, ребра, позвонки - по 1-2 случая. Первыми признаками при моносистемном ГКЛ отмечены болевой синдром - 18 пациентов (100%) и наличие опухолевидного образования у 11 детей (61,1%). Первоначально 13 детей (72,2%) были госпитализированы в хирургические отделения с подозрением на остеомиелит или опухоль кости. На рентгенограмме пораженной кости обнаружены участки просветления с утолщением кортикального слоя; на МРТ, КТ выявлены очаги деструкции костной ткани размерами от 0,5 до 6,0 см, замещенные мягкотканым компонентом (рис. 2). В биоптатах пораженной ткани находили большое количество эозинофилов и клетки Лангерганса. Трое детей с небольшим одиночным очагом деструкции лечение не получали, 10 пациентов (55,5%) оперированы: секвестрэктомия, резекция кости, удаление мягкотканого компонента. Химиотерапия проведена 7 (38,9%) детям: с полифокальным процессом - 4, с одним большим очагом деструкции - 3. Использовались международные протоколы лечения ГКЛ: LCHII, LCHIII, DAL-HX-83. Положительный эффект достигнут у всех леченых детей (100%). Тугоухость как осложнение развилась у 1 ребенка при ГКЛ с поражением левой височной кости.
Полисистемный вариант ГКЛ установлен 7 детям в возрасте от 9 мес. до 2-х лет. Болезнь дебютировала астеническим синдромом (вялостью, слабостью, беспокойством, снижением аппетита), длительной фебрильной лихорадкой у 4-х детей, увеличением лимфатических узлов в виде конгломератов у 2-х. Поражение кожи наблюдалось у 3-х пациентов в виде геморрагической петехиальной сыпи, себорейного дерматита волосистой части головы и изъявления естественных складок. Гепатоспленомегалия отмечалась у 3-х (значительно увеличивались размеры селезенки - нижний край определялся в малом тазу), у одного ребенка развился синдром несахарного диабета. В анализах периферической крови в 4-х случаях выявлялся выраженный нейтрофильный лейкоцитоз до 30 тыс/мкл со сдвигом влево, тяжелая анемия, требовавшая заместительной терапии, тромбоцитопения, ускорение СОЭ до 30мм/час. В тяжелом состоянии госпитализировано 6 больных. Диагноз мультисистемный ГКЛ установлен на основании гистологического исследования пораженной ткани (кожи, лимфоузла, кости, селезенки), КТ, МРТ, миелограммы. Хирургическое лечение, спленэктомия, поведено 1 ребенку, в катамнезе без осложнений и рецидива заболевания. Химиотерапию получили 5 детей: протокол DAL-HX-83 (2), протокол LCHII(3). В РДКБ (г.Москва) направлен 1 ребенок с полисистемным ГКЛ: поражение кожи, лимфоузлов, печени, селезенки, легких, костного мозга, множественные костные деструкции; положительный эффект достигнут после блоков химиотерапии с применением кладрибина, цитозара, солу-медрола; в настоящее время на поддерживающей терапии. В катамнезе у 6 детей с полисистемным ГКЛ отмечены осложнения: несахарный диабет (3), компрессионный перелом позвоночника (2) (рис. 4), неполная клиническая ремиссия (2).
Заключение. ГКЛ является достаточно редким заболеванием, выявляемость ГКЛ от 1 до 4-х первичных больных в год, чаще болеют мальчики. При моносистемном ГКЛ преобладает поражение костной системы, течение - благоприятное. Полисистемный ГКЛ наблюдается у детей раннего возраста; тяжесть состояния и прогноз зависят от поражения «органов риска». При этом варианте ГКЛ эффективность лечения по стандартным протоколам химиотерапии оказалась не достаточной, положительный эффект достигнут у одного больного при использовании в лечении кладрибина в сочетании с цитозаром и солу-медролом.
Решение многих проблем диагностики и лечения ГКЛ, как относительно редкого заболевания, возможны при совместных клинических исследованиях. Участие в кооперированном исследовании протокола лечения ГКЛ дало бы возможность улучшить результаты терапии пациентов с ГКЛ.
Литература:
1. Гистиоцитозы детского возраста / М. Л. Минков, Г. А. Новичкова, Г. Цельгер [и др.]. - М. : МАКС Пресс, 2005. - 156 с.
2. Махонова, Л. А. Гистиоцитарные заболевания у детей / Л. А. Махонова, Л. А. Дурнов Л. А. - М. : Медицинское информационное агенство, 2004. - 103 с.
3. Минков, М. Гистиоцитоз из клеток Лангерганса: результаты кооперированных исследований / М. Минков, Х. Гаднер // Вопросы гематологии / онкологии и иммунопатологии в педиатрии. - 2004. - т. 3, № 3. - С. 7-10.
Рис. 1. Боковая краниограмма, больной Б. С., 3 года. Множественные литические очаги деструкции в костях черепа.
Рис. 2.Компьютерная томография черепа больной Х.В., 6 лет. Одиночный очаг деструкции лобной кости с мягкотканым компонентом.
Рис. 3.Рентгенограмма правой голени больного С.В., 5 лет с диагнозом ГКЛ. Очаг деструкции с отеком и инфильтрацией в средней и нижней трети правой большеберцовой кости.
Рис. 4.Больной Б.С., 14 лет. МРТ позвоночного столба. Компрессионный перелом тел Th2, Th5, Th6, Th8, Th12 позвонков Iстепени на фоне ГКЛ.
Из литературных источников.
https://www.ajronline.org/doi/full/10.2214/AJR.09.2964
Интрадуральная эозинофильная гранулема
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5652098/
Рисунок 1
Гистоцитоз из клеток Лангерганса
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5595605/
Atlanto-axial langerhans cell histiocytosis in a child presented as torticollis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5561504/
Первоначальное изображение
Дифференциальная диагностика гранулематозных заболеваний легких: подсказки и подводные камни
http://err.ersjournals.com/content/26/145/170012
Eosinophilic granuloma at the cerebellopontine angle in an adult; a rare case report and literature review
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5508616/
Исход эозинофильной гранулемы
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5484214/
+1