(Новости лучевой диагностики 1998 4: 38)
Черепно-ключичный дизостоз.
Криводуд И. В, Другак Е. В.
БСМП, ТМО “Фтизиатрия” г. Бобруйск.
Черепно-ключичный дизостоз (dysostosis cleidocranialis congenita) относится к смешанным формам системных заболеваний скелета и впервые описан J. K. Martin в 1765 г. Подробнее заболевание позднее было изучено Barloy, Мари и Сентоном. Этиология и патогенез неизвестны. Наследуется по аутосомно-доминантному типу. В мировой литературе описано более 400 случаев данной патологии.
Наиболее характерными клинико-рентгенологическими проявлениями черепно-ключичного дизостоза являются специфические врожденные изменения ключиц. Полное отсутствие ключиц — редкое явление (10%). Обычно отсутствует акромиальный отдел или ключица фрагментирована, лопатки уменьшены в размерах. Изменён череп: увеличена мозговая и уменьшена лицевая части. Наблюдаются изменения свода черепа: в местах перекрёста швов — фонтанеллы, большой родничок может оставаться открытым в течение всей жизни. Среди швов могут быть добавочные включения — вормиевы косточки. Верхняя челюсть с придаточными полостями недоразвиты, уменьшены в объёме, твёрдое нёбо укорочено. Характерно запоздалое и несовершенное развитие постоянных зубов (молочные зубы задерживаются до 25-30 лет). По этой причине пациенты часто лечатся у стоматолога. Неправильность развития ключиц может повлечь за собой сдавление нервного плечевого сплетения, а также общую мышечную слабость верхних конечностей. Могут поражаться шейка бедра со вторичной варусной деформацией, лонная кость и другие элементы таза, в частности крестец. Иногда изменяются фаланги — укорочены дистальные части, особенно первых пальцев рук и ног с отсутствием ногтевого бугорка, а также непропорционально длинные вторые пястные и плюсневые кости. Иногда не срастаются дужки позвонков.
Больная С., 50 лет, поступила в пульмонологическое отделение с диагнозом двухсторонней пневмонии. Жалобы на одышку, боль в грудной клетке справа, повышение температуры до 38 градусов, сухой кашель. Лечилась амбулаторно без эффекта.
Из анамнеза известно, что мсать во время беременности болела тифом (каким не известно). Об отсутствии ключиц и грудины знает с детства.
Грудная клетка обычной формы, при дыхании усиленна экскурсия передней грудной стенки, визуализируется вздрагивание грудной клетки от пульсации сердца. Грудина и ключицы пальпаторно не определяются. Может свести плечи вместе и обнять грудную клетку.
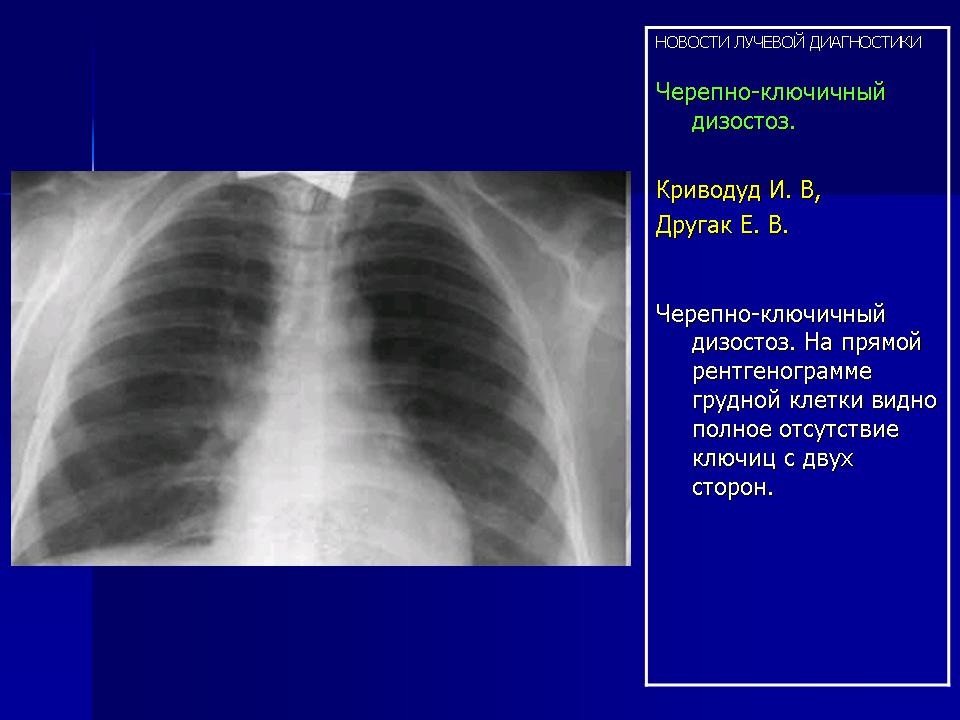
Выполнены обзорная рентгенограмма легких (Рисунок)и рентгенограмма грудины в передней косой проекции. Определяются полная агенезия грудины и ключиц с гипоплазией и деформацией лопаток.
Таким образом, описываемый случай отличителен полным отсутствием ключиц и грудины с недоразвитием лопаток. Остальные части скелета не изменены.
Черепно-лицевой дизостоз Крузона, или «попугайная» болезнь, - краниостеноз, обусловленный сочетанием недоразвития костей черепа и преждевременным зарастанием черепных швов. Проявляется изменением формы мозгового и лицевого черепа, при этом характерны гипертелоризм, экзофтальм, страбизм, своеобразная крючковатая форма носа, напоминающего клюв орла или попугая. Возможны недоразвитие нижней челюсти, нарушение прикуса: нижние зубы впереди верхних (прогнатия), снижение слуха, пирамидная и мозжечковая недостаточность, реже - другие очаговые неврологические симптомы. Могут быть различные аномалии костей туловища и конечностей. На глазном дне нередко отмечаются признаки застоя, который может смениться вторичной атрофией дисков зрительных нервов, сопровождающейся нарушением зрения.
Наследуется по аутосомно-доминантному типу. Описал в 1912 г. французский врач О. Crouzon (1874-1938).
Черепно-лицевой дизостоз Франческетти-Цвалена характеризуется грубыми нарушениями строения мозгового и лицевого отделов черепа («рыбье лицо»). Лицо вытянуто, разрез глаз антимонголоидный, верхняя и нижняя челюсти с обеих сторон недоразвиты, отмечаются гипоплазия структур пирамид височных костей, деформации ушных раковин, выраженное снижение слуха, иногда вплоть до глухоты. Нередко сочетается с другими пороками развития. Наследуется по аутосомно-доминантному типу.
Черепно-ключично-тазовый дизостоз Шенте-Мари-Сентона - семейное заболевание, характеризующееся запаздывающим зарастанием черепных швов и родничков, брахицефалией, выраженным гипертелоризмом, гиперостозом дна средней черепной ямки, отсутствием пневматизации пирамид височных костей, недоразвитием верхних челюстей и гайморовых пазух, запоздалым развитием и дистрофией постоянных зубов, частичным или полным недоразвитием ключиц (вследствие чего плечевые суставы можно сближать на груди до их соприкосновения), сколиозом, глубоким поясничным лордозом, иногда расщеплением дужек позвонков, спинномозговыми грыжами. Возможны проявления сдавливания плечевых сплетений. Грудная клетка конической формы, таз узкий, позднее окостенение лобковых костей, брахидактилия, брахиме-зофалангия, иногда прогрессирующее снижение слуха. При рентгенографии выявляют склероз костной ткани, деформации костей, множественные шпо-ровидные костные утолщения. Наследуется по аутосомно-доминантному типу. Возможны и спорадические случаи. Описали в 1898 г. J. Shentaner, P. Marie и R. Sainton.
Scheuthauer-Marie-Sainton syndrome
http://www.contempclindent.org/article.asp?issn=0976-237X;year=2012;volume=3;issue=3;spage=338;epage=340;aulast=Kuruvila
https://radiopaedia.org/articles/cleidocranial-dysostosis