
Эхографическая диагностика порока Арнольда-Киари у плода.
Научный центр акушерства, гинекологии и перинатологии РАМН,
Москва, Россия.
Материалом для данного исследования явились 55 ультразвуковых исследований у 55 беременных при сроках 15-40 недель гестации с верифицированным синдромом Арнольда-Киари у плода. Выявлен ряд особенностей в эхографическом изображении мозга при наличии этой аномалии в различные сроки гестации. Проведен изолированный анализ эхографических критериев порока в зависимости от плоскости сканирования и срока беременности. Показано снижение значимости известных эхографических признаков менингомиелоцеле у плода ("банан", "лимон") с увеличением срока беременности. Предложены новые, по мнению автора, более информативные критерии порока Арнольда-Киари, выявляемые в горизонтальных плоскостях (затрудненная визуализация мозжечка, расширение надшишковидного кармана 3-го желудочка, особая форма задних отделов тела бокового желудочка, а также вентрикуломегалия без расширения передних отделов 3-го желудочка). Указывается на особую информативность сагиттальной плоскости сканирования, позволяющей в 95% случаях выявлять основной морфологический признак аномалии Арнольда-Киари - вклинение мозжечка в большое затылочное отверстие и удлинение ствола мозга. Показана исключительная значимость исследования головного мозга у плода для выявления Spina Bifida.
Введение
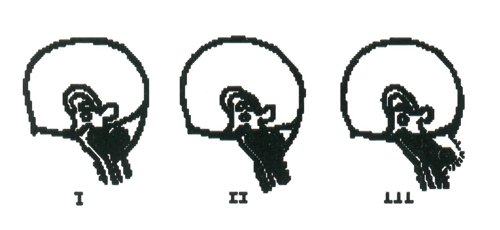
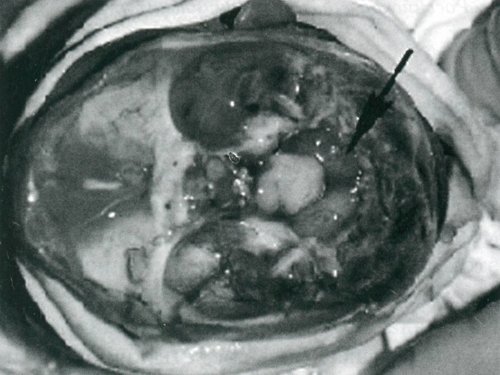
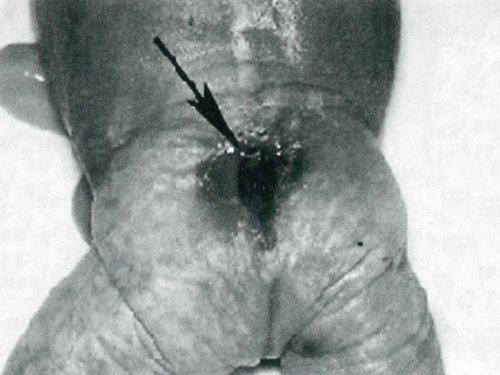
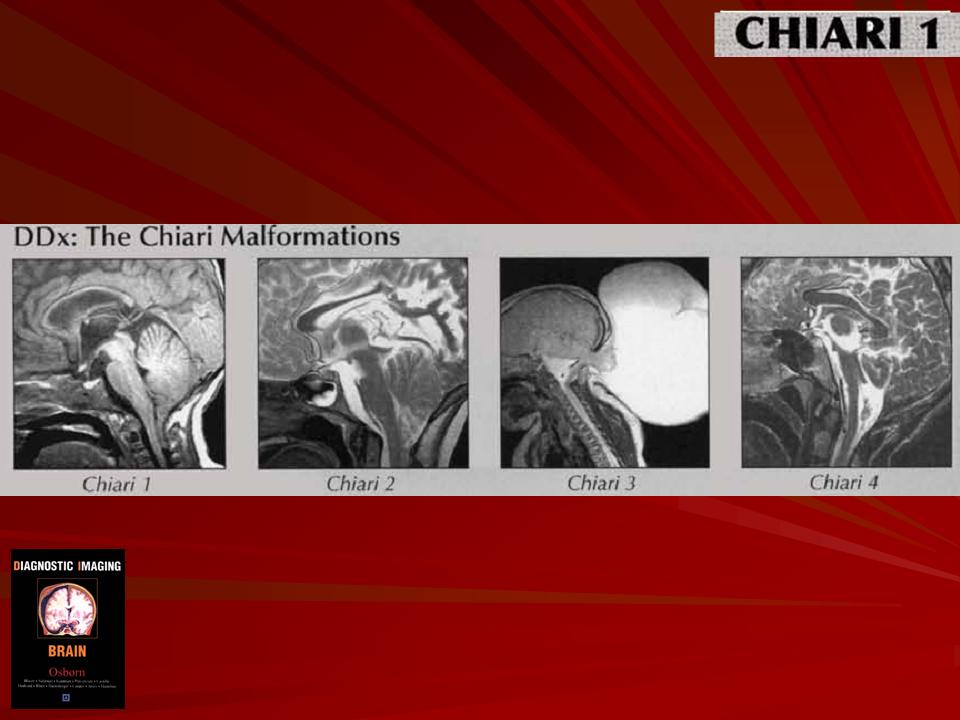
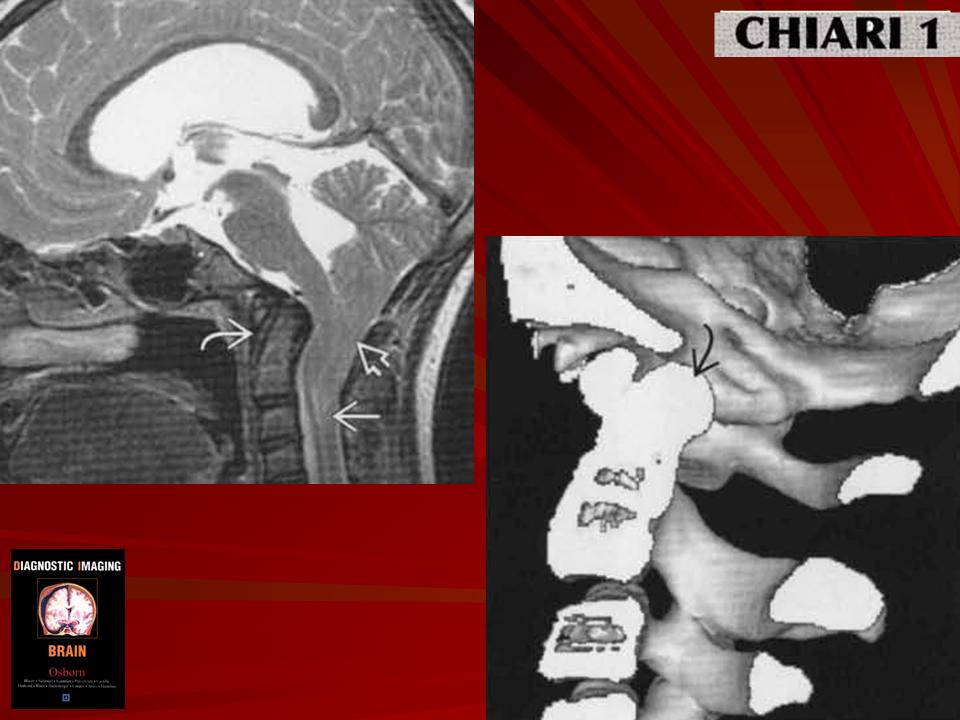
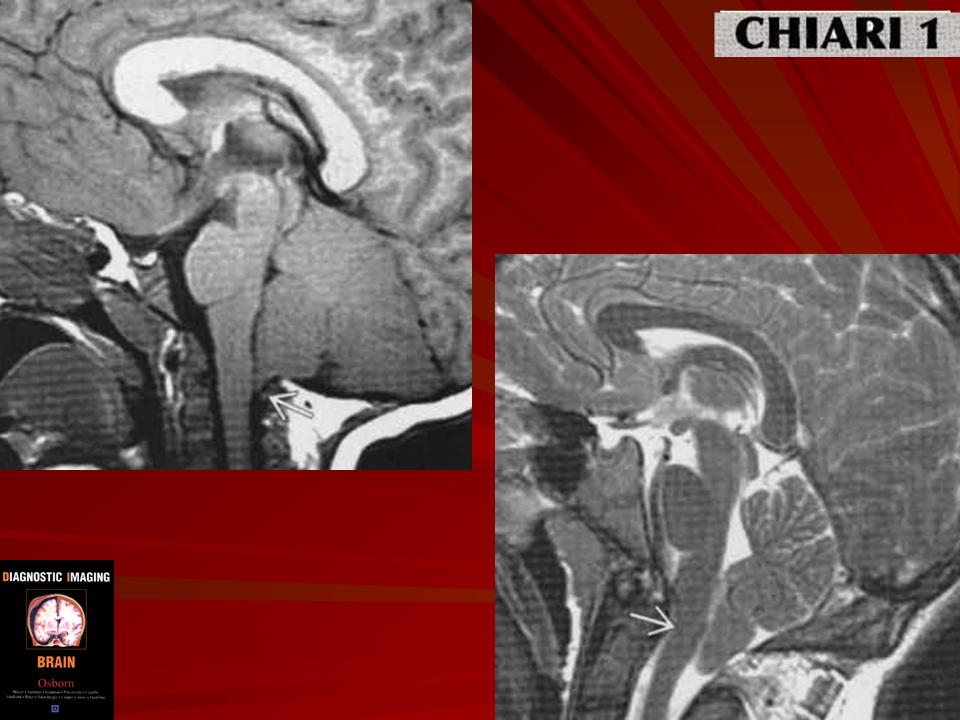
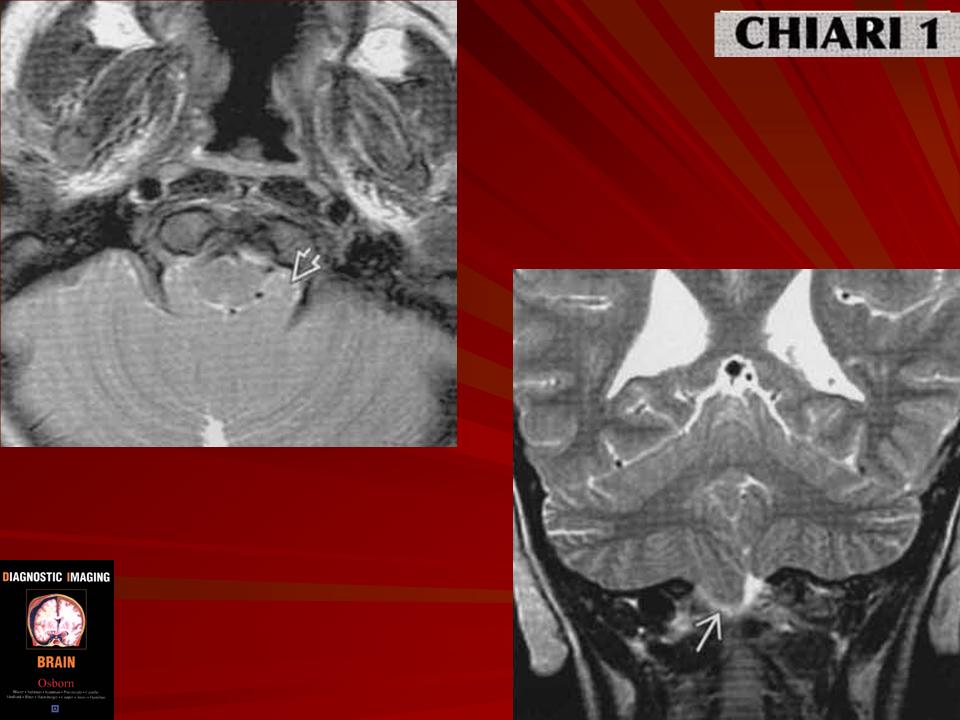
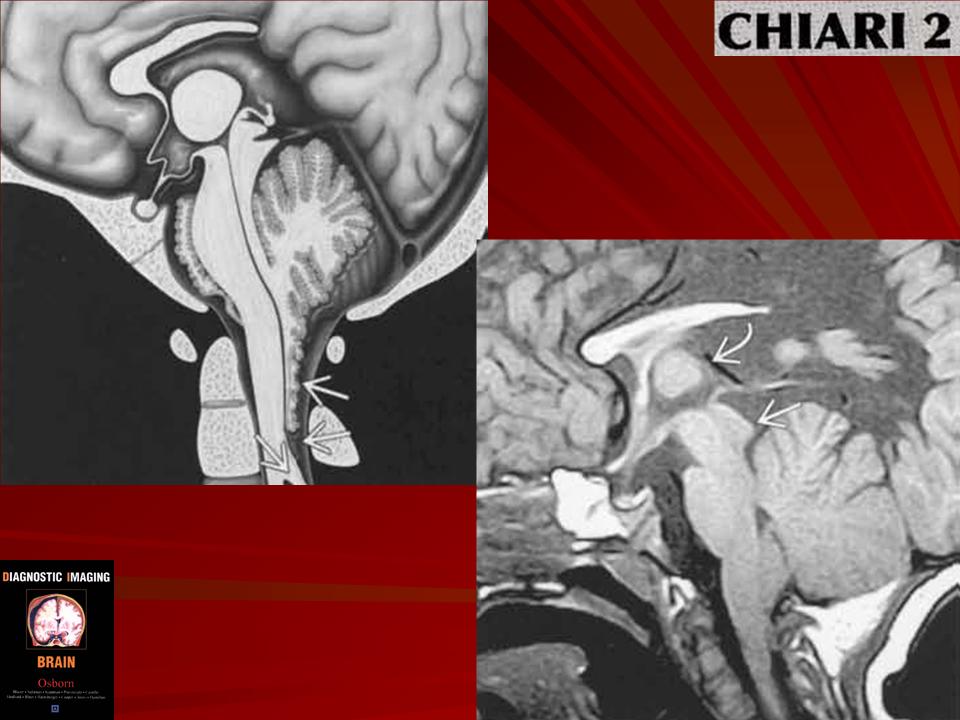
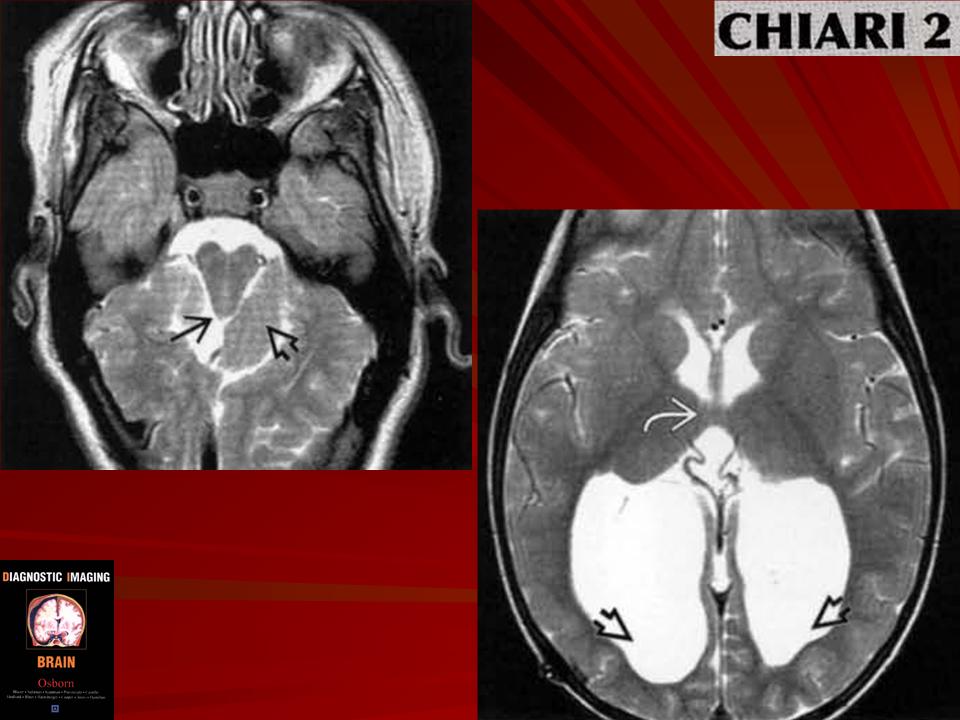
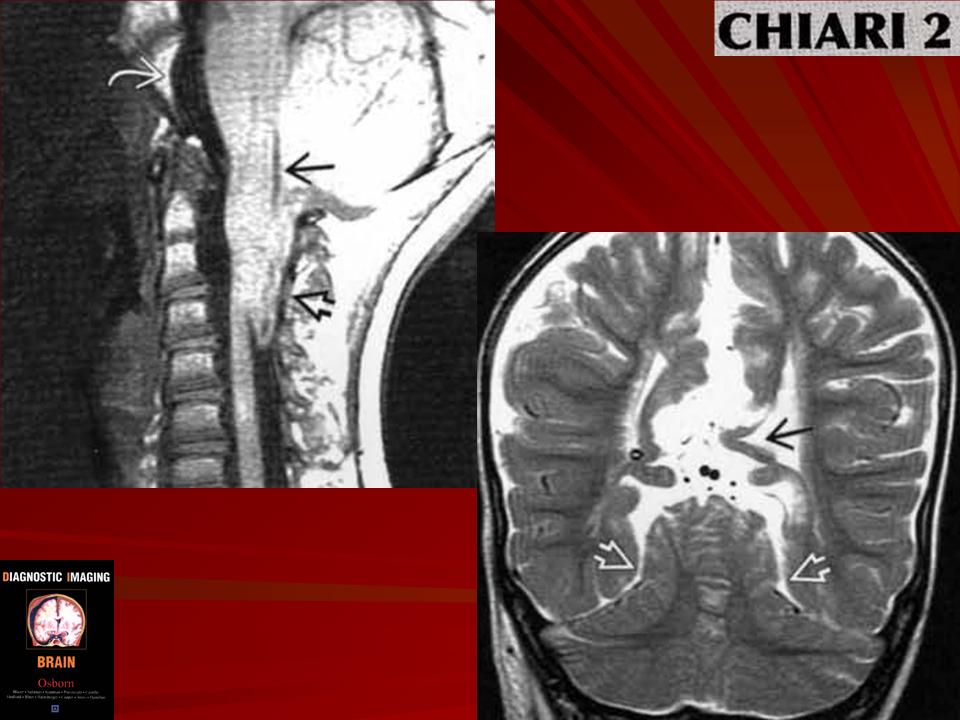
Аномалия развития головного мозга - порок (синдром) Арнольда-Киари впервые был описан в 1986 г. [1]. Современная патоморфология выделяет три основные типа этой аномалии: I - проникновение миндалин мозжечка в шейный отдел позвоночного канала; II - вклинение дисплазированного мозжечка в большое затылочное отверстие в сочетании с удлинением ствола мозга; III - изолированное тотальное смещение структур заднего мозга в расширенное затылочное отверстие, сопровождаемое образованием грыжи (рис. 1). I тип обычно не сопровождается поражением спинного мозга и выявляется чаще у взрослых при помощи КТ и ЯМР [2]. II и III типы порока характеризуются высокой летальностью в перинатальном периоде или раннем детском возрасте [2, 3]. По данным аутопсии у детей с менингомиелоцеле аномалию Арнольда-Киари II типа обнаруживают в 95-100% случаев [2, 3] (рис. 2, 3).
Точная частота синдрома Арнольда-Киари неизвестна, однако менингомиелоцеле встречается в 1-4 случаях на 1000 рождений [4, 5], занимая одно из первых мест в структуре аномалий ЦНС. Отрицательные результаты в попытках лечения детей с менингомиелоцеле, возможно, объясняются наличием дизонтогенеза головного мозга [2]. В связи с этим возрастает роль пренатального определения не только грыжи спинного мозга, но и оценки развития головного мозга у плода с целью выявления или исключения порока Арнольда-Киари.
В большинстве публикаций как отечественных, так и зарубежных авторов, посвященных уьтразвуковой диагностике пороков ЦНС у плода, отмечаются значительные сложности в выявлении менингомиелоцеле, особенно во II триместре беременности [6-8]. При упоминании об аномалии Арнольда-Киари также обычно указывается на значительные трудности в типировании и диагностике порока [8, 9]. На современном этапе к возможным эхографическим признакам аномалии Арнольда-Киари относят внутреннюю гидроцефалию [8, 9], а также типичную форму мозгового черепа типа "лимон" и изображение мозжечка в виде "банана" [4, 9]. Однако по мнению некоторых исследователей эти признаки не отличаются высокой специфичностью по отношению к пороку Арнольда-Киари [7]. Целью настоящего исследования являлось определение надежных ультразвуковых критериев диагностики аномалии Арнольда-Киари у плода, а также разработка рациональных методических приемов сканирования при подозрении на этот порок.
Материалы и методы
Материалом для настоящего исследования явились 55 ультразвуковых исследований у 55 беременных при сроках 15-40 недель гестации с верифицированным синдромом Арнольда-Киари у плода. Возраст матерей колебался от 18 до 38 лет (средний возраст 26,4 года). Возраст отцов варьировал от 21 до 46 лет (средний возраст 29,6 года). В 23 случаях (41 %) пациентки являлись первобеременными. Повторнобеременные первородящие - 5 случаев. Повторнородящие составили 49,1% (27 человек), из них у 9 детей в анамнезе отмечены пороки развития ЦНС (анэнцефалия, Spina Bifida, синдром Смита-Лемли-Опитца, гидроцефалия). У 18 женщин в анамнезе имелись здоровые дети.
После проведенного эхографического исследования в 50 случаях беременность была прервана по медицинским показаниям в сроках 20-34 недели беременности. У 5 женщин беременность закончилась рождением живого ребенка с менингомиелоцеле. Во всех случаях живорождения дети умерли в возрасте от 12 суток до 5 месяцев жизни. Во всех наблюдениях произведено патологоанатомическое исследование. Spina Bifida отмечена в 54 случаях, из них пояснично-крестцовая локализация составила 52 (94,5%), шейная - 3 наблюдения (5,5%). Внутренняя гидроцефалия обнаружена у 44. У 2 плодов отмечена агенезия мозолистого тела, у 1 косолапость, диафрагмальная грыжа и атрезия кишечника, у 2 - микроцефалия, у 2 - наружная гидроцефалия, и у 3 - порэнцефалические кисты головного мозга. Аномальное развитие задней черепной ямки и мозжечка, характерное для порока Арнольда-Киари, отмечено во всех наблюдениях.
Эхографическое обследование плодов производилось при помощи различных ультразвуковых приборов с использованием как трансабдоминального, так и трансвагинального доступов датчиками частотой 3,5-7,5 МГц. После проведения рутинного акушерского ультразвукового обследования у всех плодов производилось сканирование головного мозга с использованием трех взаимно перпендикулярных плоскостей (горизонтальной, фронтальной, сагиттальной), при этом в каждой плоскости отмечались особенности эхографической картины.
Результаты
Для выяснения возможного разнообразия эхографического изображения мозга у плодов с пороком Арнольда-Киари в различные сроки беременности с использованием трех плоскостей сканирования все обследования были разделены на три группы: 1 - плоды при беременности 15-22 недели гестации (n=18), 2 - 23-28 недель (n=17), 3 - 28-40 недель (n=20). Выявленные особенности в эхографическом изображении мозга у плодов с синдромом Арнольда-Киари при использовании горизонтальной, фронтальной и сагиттальной плоскостей в отдельных группах представлены в табл. 1-3.
При проведении эхографического обследования спинного мозга Spina Bifida была обнаружена у 53 плодов (у 3 - шейной локализации, у 50 - пояснично-крестцовой локализации). Размер дефекта колебался от 1 до 5 см в длину. При этом зависимости между размерами дефекта спинного мозга и эхографическим изображением головного мозга не выявлено. В 1 случае дефект позвоночника у плода в 17 недель беременности не был обнаружен из-за выраженного ожирения пациентки и расположения плода в заднем виде. Учитывая особенности анатомии задней черепной ямки, а также степень удлинения ствола мозга, исследование показало возможность диагностики типа порока Арнольда-Киари.
Порок Арнольда-Киари II типа обнаружен у 52,1 типа - у 2 и III типа - у 1 плода (рис. 4-6). При этом наиболее информативной являлась сагиттальная плоскость. Точность эхографического выявления типа порока во всех наблюдениях совпала с патологоанатомическими данными. При диагностике III типа порока Арнольда-Киари основным критерием, наряду с выраженным удлинением ствола мозга, было выявление грыжи в затылочной области, которая была сформирована за счет расширения большого затылочного отверстия. При обследовании плодов с наличием I типа синдрома выраженность удлинения стволовой части головного мозга была незначительной, обращало на себя внимание лишь отсутствие большой цистерны и субарахноидального пространства в задней части верхних отделов позвоночного канала. Диагностика II типа порока была наиболее доступной, благодаря выраженному удлинению стволовой части мозга, а также отсутствию изображения большой цистерны и субарахноидального пространства верхней части шейного отдела позвоночного канала.
В результате эхографических обследований головного мозга плодов с пороком Арнольда-Киари в горизонтальных плоскостях (см. табл. 1) вентрикуломегалия, а именно увеличение ширины задних отделов тела бокового желудочка, обнаружена в 41 случае. Следует отметить, что расширение тел боковых желудочков в наших исследованиях не всегда сопровождалось расширением 3-го желудочка при сроках беременности 36-40 недель. Во всех наблюдениях расширение передних отделов 3-го желудочка сочеталось с расширением ширины задних отделов тела бокового желудочка более 2 см.
Признак порока (особенность)Число случаев и процент к числу наблюдений в группах1
(n=18)2
(n=17)3
(n=20)"Банан"3 (16,6%)2 (11,7%)0 (0%)"Лимон"5 (27,7%)2 (11,7%)0 (0%)"Банан" + "Лимон"3 (16,6%)0 (0%)0 (0%)Вентрикуломегалия
(ширина задних отделов тела бокового желудочка > 1,0 см)11 (61,0%)13 (76,4%)17 (85,0%)Расширение 3-го желудочка:
передних отделов
задних отделов
0 (%)
9 (50,0%)
1 (5,8%)
7 (41,1%)
5 (25,0%)
10 (50,0%)Ланцетовидная форма задних отделов тела желудочка8 (44,0%)7 (41,1%)11 (55,0%)Затрудненная визуализация мозжечка12 (66,0%)15 (88,2%)19 (95,0%)
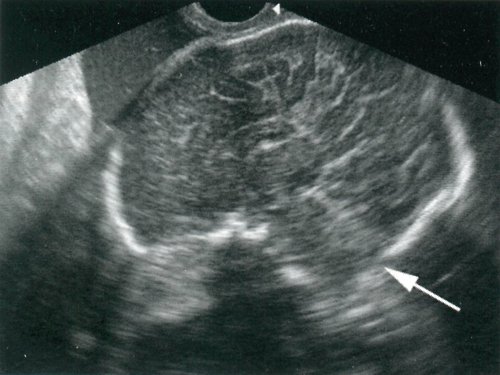
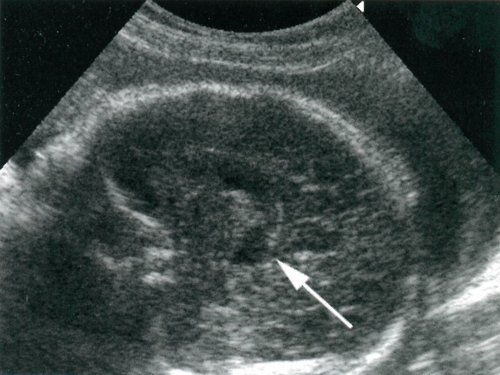
Расширение задних отделов 3-го желудочка в настоящем исследовании обнаружено у 26 плодов. Выявление ромбовидной структуры в области цистерны четверохолмия (см. рис. 5), соответствующее расширению надшишковидного кармана 3-го желудочка, сочеталось либо с полным отсутствием гидроцефалии, либо с вентрикуломегалией, когда ширина задних отделов тела бокового желудочка была менее 2 см. Следует отметить, что расширение задних отделов 3-го желудочка у плодов с пороком Арнольда-Киари отмечено в половине случаев в каждой из групп (см. табл. 1).
Без четкой корреляции со сроком беременности у плодов с пороком Арнольда-Киари при горизонтальных сканированиях выявлена характерная, заостренная кзади ("ланцетоподобная"), форма задних отделов тела бокового желудочка, которая в абсолютном числе наблюдений коррелировала с характерными изменениями формы боковых желудочков при фронтальных сканированиях.
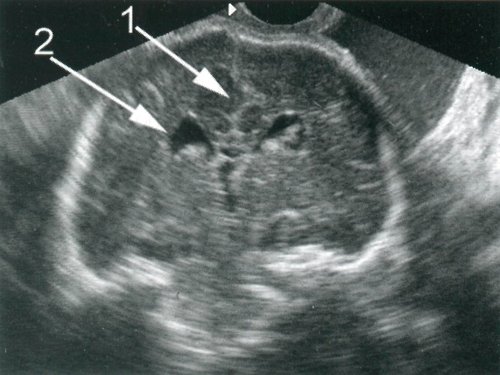
При обследованиях мозга плодов во фронтальных плоскостях тело бокового желудочка на протяжении от отверстия Монро до треугольника приобретало дополнительную латеральную стенку. Следует отметить тенденцию к учащению появления этого признака у плодов с увеличением гестационного срока (см. табл. 2). Подобная тенденция наблюдалась также в эхографическом выявлении асимметрии расположения сосудистых сплетений боковых желудочков (рис. 6). Искривления мозолистого тела и асимметрия борозд медиальных поверхностей полушарий чаще отмечались у плодов во 2 и 3 группах. Отсутствие прозрачных перегородок не коррелировало с гестационным возрастом.
Признак порока (особенность)Число случаев и процент к числу наблюдений в группах1
(n=18)2
(n=17)3
(n=20)Атипичность субарахноидальных пространств лобных долей6 (33,3%)13 (76,4%)16 (80,0%)Изменение формы боковых желудочков8 (44,4%)12 (70,5%)19 (95,0%)Асимметрия расположения сосудистых сплетений боковых желудочков11 (61,1%)13 (76,4%)15 (75,0%)Искривление молозистого тела и асимметрия борозд медиальных поверхностей полушарий3 (16,6%)5 (29,4%)9 (45,0%)Отсутствие прозрачной перегородки2 (11,1%)3 (17,6%)3 (15,0%)
При фронтальных сканированиях через лобные доли у плодов с синдромом Арнольда-Киари во 2 и 3 группах (см. табл. 2) выявлено частое атипичное изображение латеральных субарахноидальных пространств лобных долей, которое проявлялось в резком сужении или даже в полном их отсутствии при сроках беременности менее 34 недель.
Резкое удлинение ножек мозга и моста, отмеченное при сагиттальных сканированиях у плодов с пороком Арнольда-Киари, в подавляющем большинстве случаев (см. табл. 3) характеризовалось увеличением расстояния (2 см или более) между задней поверхностью валика мозолистого тела и верхней поверхностью червячка мозжечка (см. рис. 5). Следует отметить, что при наличии порока Арнольда-Киари I типа удлинение ножек мозга и моста было незначительным (см. рис. 4). Во всех случаях диагностики порока Арнольда-Киари I типа выявлялось, прежде всего, отсутствие большой цистерны, а также отмеченные выше другие признаки этой аномалии.
Признак порока (особенность)Число случаев и процент к числу наблюдений в группах1
(n=18)2
(n=17)3
(n=20)Отсутствие большой цистерны вследствие смещения мозжечка12 (66,6%)16 (94,1%)19 (95,0%)Резкое удлинение ножек мозга и моста17 (94,4%)15 (88,2%)19 (95,0%)
Обсуждение
Ультразвуковая диагностика пороков развития ЦНС у плода за последнее десятилетие значительно повысила свои потенциальные возможности [13]. По данным Nelson N. L. et al. [10], открытые формы спинномозговой грыжи могут быть диагностированы у плода в 88% случаев, а закрытые формы - в 76%. Однако эти же авторы подчеркивают, что абсолютная возможность ультразвукового определения Spina Bifida во время пренатальных обследований колеблется от 22 до 0%. По-видимому, такая ситуация в диагностике одного из наиболее распространенных пороков ЦНС у плода обусловлена сложившейся к настоящему времени общей системой подхода к выявлению этой аномалии, включающей в себя эхографическое обнаружение признаков "лимона" и "банана", а также вентрикуломегалии в горизонтальной плоскости в сочетании с продольным и поперечным визуальным исследованием позвоночника.
Анализируя результаты настоящего исследования (см. табл. 1), при использовании горизонтальных плоскостей сканирования совокупная выявляемость таких признаков порока Арнольда-Киари, как "лимон" и "банан", составила не более 45% при сроке гестации до 22 недель и 24% при беременности до 28 недель. Вентрикуломегалия (увеличение ширины задних отделов тела бокового желудочка или atrium более 1,0 см) встретилась в 61, 76 и 85% (в 1,2 и 3 группах соответственно). Однако по данным Американского института ультразвука в медицине вентрикуломегалия при ширине задних отделов тела бокового желудочка более 1,5 см абсолютно точно устанавливается лишь в 53% случаев [10]. Затруднительное выявление или полное отсутствие визуализации мозжечка в сочетании с выявлением "ланцетовидной" формы задних отделов тела боковых желудочков и расширения надшишковидного кармана 3-го желудочка, по нашему мнению, по своей информативности значительно превосходят другие критерии порока Арнольда-Киари и менингомиелоцеле при горизонтальных сканированиях (см. табл. 1). Достаточную помощь в постановке диагноза порока Арнольда-Киари может также оказать определение вентрикуломегалии с отсутствием признаков расширения передних отделов 3-го желудочка. На это указывают как данные настоящего исследования, так и результаты ультразвуковых обследований у новорожденных с менингомиелоцеле [12].
Отсутствие в литературе данных по применению фронтальных и сагиттальных плоскостей сканирования у плодов с Spina Bifida или синдромом Арнольда-Киари несколько затрудняет интерпретацию результатов, полученных в ходе настоящего исследования (см. табл. 2, 3). Однако они не противоречат как патоморфологическим представлениям о макроскопической организации порока, так и данным постнатальных исследований мозга с применением различных визуальных методов [2, 3,11,12].
Результаты эхографических обследований, приведенные в табл. 2, указывают на наличие ряда особенностей мозга, которые могут быть выявлены при использовании фронтальных сканирований у плодов с менингомиелоцеле. Несмотря на высокую выявляемость (см. табл. 2) таких признаков, как атипичность субарахноидальных пространств лобных долей, изменение формы боковых желудочков и асимметрию сосудистых сплетений и борозд, эти критерии, по-видимому, отражают больше вторичные общие дизонтогенетические изменения мозга, связанные с различными вариантами аномального развития ЦНС [14]. Вместе с тем выявление таких особенностей может способствовать более точной диагностике аномального головного мозга и дополнять картину прогноза в каждом конкретном случае.
Частота эхографической выявляемости таких признаков, как отсутствие большой цистерны и удлинение ножек мозга (см. табл. 3), убедительно показала преимущества сагиттальной плоскости сканирования мозга для диагностики основного макроскопического признака порока Арнольда-Киари - вклинения частей мозжечка в большое затылочное отверстие. Получение изображения головного мозга плода в сагиттальной плоскости во II триместре беременности, по нашему мнению, не является сложной технической задачей. В связи с этим использование сагиттальной плоскости сканирования мозга можно предложить как одну из наиболее информативных плоскостей для диагностики или исключения аномалии Арнольда-Киари у плода как во II, так и в III триместрах беременности.
Обнаружение признаков аномалии Арнольда-Киари у плода может явиться показанием к исключению дефектов развития позвоночника (включая инвазивные), при этом даже полное отсутствие ультразвуковой информации о состоянии позвоночника указывает на наличие Spina Bifida более чем в 95% случаев.
Таким образом, выполненное исследование показало, что использование не только горизонтальной, но и других плоскостей сканирования (фронтальной, сагиттальной), позволяет выявить ряд высокоинформативных критериев аномалии Арнольда-Киари у плода. Эхографический диагноз этого порока при использовании совокупности плоскостей и учета перечисленных выше признаков не должен представлять затруднений в подавляющем большинстве случаев после 16-й недели гестации.
Аномалия Арнольда - Киари (Chiari malformation)
Частота этого заболевания составляет от 3.3 до 8.2 наблюдений на 100000 населения.
До настоящего времени патогенез патологии окончательно не установлен. По всей вероятности, этих патогенетических факторов три: первый - наследственно обусловленные врожденные остеоневропатии, второй - травматические повреждения клиновидно-решетчатой и клиновидно-затылочной части ската вследствие родовой травмы, третий - гидродинамический удар ликвора в стенки центрального канала спинного мозга.
В 1891 году Киари ( Chiari ) выделил четыре типа аномалии с подробным их представлением. Данной классификацией мы пользуемся по настоящее время.
- Аномалия Арнольда-Киари I типа представляет собой опущение структур ЗЧЯ в позвоночный канал ниже плоскости большого затылочного отверстия.
- При аномалии Арнольда-Киари II типа - происходит каудальная дислокация нижних отделов червя, продолговатого мозга и IV желудочка, нередко развивается гидроцефалия.
- Аномалия Арнольда-Киари III типа встречается редко, характеризуется грубым каудальным смещением всех структур задней черепной ямки.
- Аномалия Арнольда-Киари IV типа - гипоплазия мозжечка без смещения его вниз.
Аномалии III и IV типов обычно несовместимы с жизнью.
Примерно у 80% пациентов АК сочетается с патологией спинного мозга - сирингомиелией , которая характеризуется образованием в спинном мозге кист, вызывающих прогрессирующую миелопатию. Эти кисты образуются при опущении структур ЗЧЯ и сдавление шейного отдела спинного мозга.
Типичная клиническая картина аномалии Арнольда-Киари характеризуется следующими симптомами:
- боль в шейно-затылочной области усиливающаяся при кашле, чихании,
- снижение болевой и температурной чувствительности в верхних конечностях,
- снижение мышечной силы в верхних конечностях,
- спаситичность верхних и нижних конечностей,
- обмороки, головокружения,
- снижение остроты зрения,
- в более запущенных случаях присоединяются: эпизоды апноэ (короткая остановка дыхания), ослабление глоточного рефлекса, непроизвольные быстрые движения глаз.
Иногда аномалия Киари никак не проявляет себя и выявляется случайно при диагностических процедурах.
В настоящий момент методом выбора при диагностике данной патологии является МРТ головного мозга шейного и грудного отделов спинного мозга (для исключения сирингомиелии).
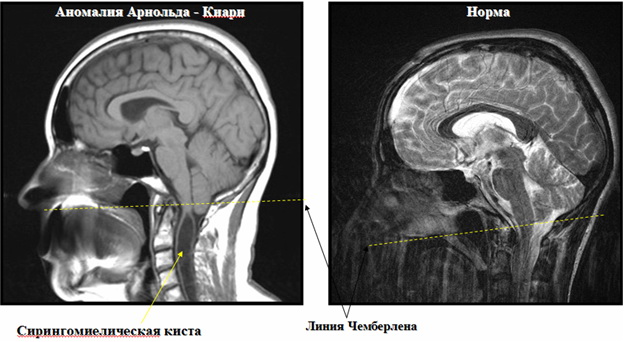
Рисунок 1. На левом снимке представлена МР картина Аномалии Арнольда-Киари I типа - опущение миндалин мозжечка ниже линии Чемберлена (линия проведенная от заднего края затылочного отверстия до твердого неба). Ниже места ущемления визуализируется сирингомиелическая киста. На правом снимке представлен вариант нормы.
Если единственным симптомом заболевания является незначительной интенсивности болевой синдром, для лечения применяется консервативная терапия, которая включает в себя различные схемы с примененим нестероидных противовоспалительных препаратов и миорелаксантов.
При отсутствии эффекта от консервативной терапии в течение 2-3 месяцев или наличии у пациента неврологического дефицита (онемение, слабость а конечностях и т.д.) показано проведение операции.
Целью операции является устранение сдавления нервных структур и нормализация тока цереброспинальной жидкости, для чего производится увеличение объема ЗЧЯ. В результате лечения, как правило, уменьшается или исчезает головная боль, частично восстанавливаются чувствительность и двигательные функции.



Синдром Арнольда - Киари - это совокупность изменений в ЦНС плода, возникающих в результате порока развития продолговатого мозга и обусловленных асинхронным ростом ствола мозга и спинного мозга. Впервые он был описан Н. Chiari в 1896 г. Это состояние характеризуется каудальным смещением продолговатого мозга, моста и червя мозжечка, когда все эти структуры оказываются в шейной части позвоночника.
Выделяют три типа мальформации Арнольда-Киари в зависимости от степени вклинения структур продолговатого мозга в позвоночник:
I тип характеризуется удлинением ствола мозга и проникновением миндалин мозжечка в шейный отдел позвоночного канала;
II тип - вклинивание дисплазированного мозжечка в большое затылочное отверстие в сочетании с удлинением ствола мозга;
III тип - тотальное смещение структур заднего мозга в расширенное затылочное отверстие, сопровождаемое грыжей в затылочной области.
Истинная частота различных типов синдрома Арнольда - Киари, да и частота этого порока в целом, не установлены. Одной из причин отсутствия таких данных являются разные подходы к классификации этого порока. Согласно Международной классификации болезней, синдром Арнольда - Киари имеет отдельный шифр (Q07.0), однако определяется в ней как «... патологическое состояние, при котором происходит повышение внутричерепного давления в результате интракраниальной опухоли, окклюзионных форм гидроцефалии, воспалительного процесса, что в некоторых случаях приводит к вклинению мозжечка и продолговатого мозга в большое затылочное отверстие». В ультразвуковой пренатальной литературе до сих пор нам не удалось найти описаний случаев дородовой диагностики синдромаАрнольда- Киари, полностью соответствующих этим характеристикам.
Морфологические особенности различных типов порока Арольда - Киари определяют возможности пренатального выявления и прогноз для жизни. I тип порока совместим с жизнью и нередко является случайной находкой у взрослых при проведении МРТ. Пороки II и III типа в перинатальном периоде встречаются достаточно часто и имеют, как правило, крайне неблагоприятные перинатальные исходы.
Среди диагностированных случаев синдрома Арнольда - Киари основная часть приходится на II тип. Так, в исследованиях СМ. Воеводина из 55 случаев пренатальной диагностики мальформации Арнольда - Киари, которые были зарегистрированы в разные сроки беременности в интервале от 15 до 40 нед I тип составил только 3,5%, II тип - 94,5%, III тип - 2%. По данным нашего центра, при анализе 29 случаев пренатального вывления изменений, которые были нами трактованы, как синдром Арнольда - Киари, лишь у одного плода была обнаружена грыжа в затылочной области, что дает возможность ретроспективно предположить наличие порока III типа (3,4%).
Причины возникновения синдрома Арнольда- Киари до конца не установлены. Хромосомные аномалии при этой патологии, как правило, выявить не удается.
Ультразвуковая диагностика синдрома Арнольда - Киари основана на оценке анатомии структур, расположенных в задней черепной ямке. В норме с конца I триместра при эхографии хорошо видны полушария и червь мозжечка, большая цистерна. В случаях изменения формы и размеров мозжечка, его нечеткой визуализации, уменьшения или исчезновения большой цистерны у врача должно возникнуть подозрение на наличие у плода синдрома Арнольда - Киари.
Очевидно, что пренатальной диагностике в основном доступны II и III тип мальформации Арнольда - Киари. I тип порока может быть заподозрен в тех случаях, когда при ультразвуковом исследовании головы плода в сагиттальной плоскости большая цистерна резко уменьшена в размерах или не визуализируется вовсе. Изображение мозжечка при этом не меняется. Основной морфологический признак этого синдрома - удлинение ствола мозга - по нашему мнению, не имеет четкихэхографическиххарактеристик и поэтому является весьма субъективным.
При II типе порока помимо отсутствия изображения большой цистерны в горизонтальной и сагиттальной плоскостях, отмечается изменение формы и положения мозжечка за счет его каудального смещения. Аномальная форма мозжечка и отсутствие привычного ультразвукового изображения большой цистерны хорошо заметны при эхографии и не вызывают трудностей у врача проводящего исследование.
При III типе порока ко всем описанным выше признакам присоединяется образование затылочной грыжи.
Существенную помощь в пренатальной диагностике синдрома Арнольда- Киари оказывает обнаружение дополнительных эхографических отклонений. В подавляющем большинстве случаев этот порок сопровождается вентрикуломегалией или гидроцефалией, которая является вторичной и легко диагностируется при ультразвуковом исследовании. Поданным отечественных авторов, увеличение ширины тел боковых желудочков мозга в горизонтальной плоскости исследования было обнаружено в 74% случаев порока Арнольда - Киари. Частота этого признака нарастала с увеличением срока беременности (61-85%). Поданным зарубежных авторов, расширение желудочков более 15 мм встречается в 53% случаев синдрома Арнольда-Киари. В наших исследованиях при синдроме Арнольда - Киари у плода изменения желудочковой системы были зарегистрированы в 89,6%.
При эхографическом исследовании плода с подозрением на синдром Арольда - Киари следует обращать внимание и на форму боковых желудочков. Иногдажелудочки меняют ее и становятся заостренными кзади, как бы «ланцетоподобными».
В некоторых случаях синдром Арнольда - Киари сопровождается изменением формы головы («лимон») часто в сочетании с аномальной формой мозжечка («банан»), однако, по мнению СМ. Воеводина, информативность этих признаков низкая. Частота встречаемости этого сочетания в ранние сроки, по его данным, составила около 45%, в интервале 22-28 нед- 24%. Аналогичные данные были приведены М. Van den Hof и соавт. В ранние сроки признак «лимон» был зарегистрирован в 98% случаев, а после 24 нед - только в 13% наблюдений. В наших исследованиях форма головы плода в виде лимона былазарегистрированатолькоу 27,6% плодов с синдромом Арнольда - Киари, при этом во всех случаях срок диагностики не превышал 24 нед.
Помимо горизонтальной и сагиттальной плоскостей в пренатальной диагностике синдрома Арнольда-Киари помощь оказывают и другие плоскости сканирования. Во фронтальной плоскости может отмечаться изменение формы боковых желудочков (70%), атипичность субарахноидальных пространств лобных долей (63%), асимметрия расположения сосудистых сплетений боковыхжелудочков (71 %). Справедливости ради следует отметить, что в практическом здравоохранении в основном используется горизонтальная и реже сагиттальная плоскости, поэтому основное внимание следует обращать на особенности визуализации структур мозжечка и размеры большой цистерны.
Очень важной особенностью синдрома Арнольда-Киари является его сочетание с аномалиями развития позвоночного столба. С одной стороны, в подавляющем большинстве случаев (до 95%) этот комплекс изменений сопровождается образованием спинномозговой грыжи. С другой стороны, патологоанатомические исследования свидетельствуют, что у детей со спинномозговой грыжей частота аномалии развития продолговатого мозга, то есть синдром Арнольда-Киари, приближается к 100%. В наших исследованиях изменения в позвоночнике мы нашли у всех плодов с подозрением на синдром Арнольда - Киари. По мнению некоторых исследователей, именно нарушения в формировании головного мозга, являются основной причиной неудачных исходов лечения спинномозговых грыж у детей.
Наличие или отсутствие спинномозговой грыжи во многом определяет прогноз при синдроме Арнольда - Киари. Учитывая высокую частоту сочетания этих двух пороков и выраженность вторичных изменений в головном мозге (вентрикуломегапия или гидроцефалия) прогноз для жизни и особенно прогноз для постнатального здоровья у плодов с наличием синдрома Арнольда - Киари можно расценивать как неблагоприятный.
Завершая раздел, посвященный вопросам пренатальной диагностики синдрома Арнольда - Киари, следует остановиться на методических особенностях, помогающих поставить точный пренатальный диагноз. Этот синдром по сути своей является пороком развития продолговатого мозга. Аномалии развития позвоночного столба часто сопровождают его, но не являются обязательной составной частью синдрома. Следовательно, пренатальный диагноз синдрома Арнольда - Киари можно ставить только в тех случаях, когда имеются явные изменения в области задней черепной ямки, при этом наличие или отсутствие нарушений строения позвоночника и изменений в желудочковой системе не являются решающими для постановки этого диагноза. Ретроспективный анализ трехлетнего периода, в течение которого были зарегистрированы 29 случаев пренатального выявления изменений у плода, определенных как синдром Арнольда - Киари, показал что в 6 из них ультразвуковые изменения мозжечка не были описаны. В 5 из 6 случаев постнатальные и/или патологоанатомические исследования сняли диагноз синдрома Арнольда - Киари. Как уже указывалось выше, наличие патологии продолговатого мозга существенно ухудшает прогноз для жизни и здоровья, поэтому точный пренатальный диагноз синдрома Арнольда - Киари играет принципиальную роль в определении тактики ведения беременности.
Аномалия Арнольда-Киари
Роль МРТ в диагностике аномалии Арнольда-Киари
Захарова Е.М.
ГУЗ «Нижегородская областная клиническая больница им. Н.А. Семашко»
Нижегородская государственная медицинская академия
Киари- 3
При мальформации И типа (Киари II) отмечается протрузия в большое затылочное отверстие миндалин и червя мозжечка, структур продолговатого мозга, который принимает S-образную форму. Характерны спастический тетрапарез, боли в затылочной области и шее, мозжечковая атаксия, вертикальный «бьющий» вниз нистагм, элементы бульбарного синдрома, признаки сирингомие-лии, проявления гидроцефалии, проводниковые нарушения.
Неврологическая симптоматика при синдроме Арнольда-Киари может проявляться с 5-7 лет, иногда позже, возможно в 30-40-летнем возрасте, и имеет прогредиентное течение. Проявления аномалии Арнольда-Киари нередко сочетаются с краниовертебральной костной аномалией (базилярная импрессия, ассимиляция атланта, краниостеноз по типу скафокрании и др.). В диагностике синдрома Киари и определении его типа особенно ценной обычно является информация, полученная при МРТ головного мозга и краниовертебральной области, а также при транскраниальной допплерографии (Крупина Н.Е., 2003).
Мальформация I типа (Киари I) характеризуется смещением миндалин мозжечка до уровня большого затылочного отверстия. Возможно опущение продолговатого мозга, его удлинение и передняя компрессия продолговатого мозга зубовидным отростком, сужение IV желудочка мозга и большой затылочной цистерны, ликвородинамические расстройства, признаки недоразвития и атипичного строения артерий вертебрально-базилярного бассейна. В неврологическом статусе возможны глазодвигательные, кохлеарные и вестибуломоз-жечковые, бульбарные, а также проводниковые двигательные и сегментарные двигательные и чувствительные нарушения. Отсутствие неврологических симптомов, однако они могут проявиться позже (иногда на 3-4 десятилетии жизни, что свидетельствует о переходе процесса в мальформацию II типа.
Детская нейрохирургия: врожденные аномалии
а. Аномалия Киари (Chiari)
Старое название аномалия Арнольда-Киари. Это патологическое состояние, связанное с неправильным формированием структур ЗЧЯ и, в ряде случаев, ствола мозга. На настоящий момент известны четыре разновидности этой патологии. Наибольшую клиническую значимость из них представляют Chiari-1 и Chiari-2.
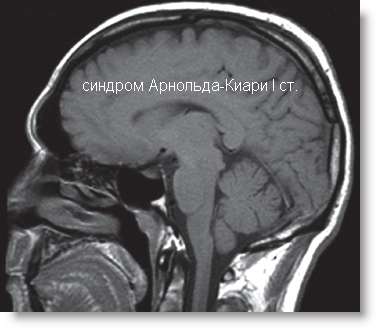
CHIARI-1 - характеризуется опущением миндаликов мозжечка ниже уровня большого затылочного отверстия, что часто сопровождается формированием сирингомиелии на шейно-грудном уровне.
Эпидемиология: средний возраст клинического проявления заболевания 40 лет, имеется незначительная предрасположенность женского пола (м/ж=1/1.3)
Основные симптомы: головные боли затылочного характера, боли по задней поверхности шеи, мозжечковая симптоматика (нарушения координации, шаткость походки), при наличии сирингомиелии характерными являются диссоциированные расстройства чувствительности на верхней половине туловища по типу "накидки". В редких случаях Киари-1 может сопровождаться окклюзионной гидроцефалией. Кроме того, следует иметь в виду, что иногда у этих больных случаются ночные остановки дыхания.
Диагностика: На настоящий момент МРТ головного мозга является методом выбора при диагностике данной патологии. Следует отметить, что кроме МРТ головного мозга при подозрении на аномалию Киари-1 необходимо сделать МРТ шейного и грудного отделов спинного мозга для исключения/подтверждения сирингомиелии. И, наоборот, при наличии сирингомиелии в обязательном порядке необходимо сделать МРТ головного мозга с особым вниманием на состояние структур ЗЧЯ.
Лечение: Асимптоматичные случаи лечения не требуют. Рекомендуется лишь контролирование ситуации путем проведения ежегодного МРТ обследования. При наличии симптоматики лечение только хирургическое. Операцией выбора является "декомпрессия области краниоцервикального перехода" с пластикой оболочки.
Исходы: Улучшение состояния наблюдается у 87% оперированных больных, стабилизация - у остальных. Риск развития серьезных осложнений составляет менее 0.5%.
CHIARI-2 - часто сопутствует миеломенингоцеле. При этом патологическом состоянии отмечается еще большее опущение миндаликов мозжечка (они обычно находятся вне полости черепа), каудальная дислокация структур ЗЧЯ - т.е. ствол, 4-й желудочек расположены значительно ниже, частично или полностью в спинномозговом канале, а каудальные черепно-мозговые нервы имеют восходящее направление (в отличие от "нисходящего" в норме).
Эпидемиология: в связи с более грубой патологией и большим вовлечением в процесс ствола мозга, заболевание проявляется в детском (часто в раннем) возрасте. Чем в более раннем возрасте происходит появление симптоматики, тем серьезнее подлежащая патология и тем хуже прогноз. У новорожденных часто бывает быстрое нарастание стволовой симптоматики с летальным исходом.
Основные симптомы: связаны с нарушением функция ствола мозга (главным образом продолговатого мозга и моста) - нарушения глотания, остановки дыхания, стридор, аспирация, тетрапарез, парез мимической мускулатуры, нарушения крика (у новорожденных)
Диагностика: МРТ является методом выбора. Кроме дистопии структур ЗЧЯ часто встречаются различные другие аномалии развития головного мозга, гидроцефалия (значительно чаще, чем при Киари-1). Ларингоскопия - помогает оценить степень нарушения иннервации глотки и гортани.
Лечение: Только хирургическое. Первым этапом необходимо разрешить проблему с гидроцефалией (шунт), далее, при сохраняющейся стволовой симптоматике производится декомпрессия ЗЧЯ и области краниоцервикального перехода с пластикой оболочки. При наличии грубых бульбарных нарушений рекомендуется трахеостомия.
Исходы: Улучшение состояния наблюдается у 80% оперированных больных (у 60% после операции наблюдается практически норма), однако у оставшихся 20% часто идет прогрессирование стволовых нарушений. Наиболее неблагоприятные исходы наблюдаются у детей до года. Основная причина летальности - остановка дыхания.
CHIARI-3 - наиболее тяжелая форма дистопии, часто имеется субокципитальное энцефаломенингоцеле. Обычно эта клиническая ситуация несовместима с жизнью.
CHIARI-4 - церебеллярная гипоплазия без дистопии миндаликов.
Статью подготовил резидент КазНМУ им. С.Д. Асфендиярова Д.В. Енцов
Некоторые формы этих пороков вызывают довольно неприятные симптомы, от простого головокружения до инсультов головного и спинного мозга. Симптоматика может долгое время отсутствовать, а затем развиться внезапно, после травмы, гриппа или другой провокации, причем в любом возрасте.
Аномалия Арнольда-Киари - это врожденная патология развития ромбовидного мозга, проявляющаяся несоответствием размеров задней черепной ямки и мозговых структур, находящихся в этой области, что приводит к опущению ствола головного мозга и миндалин мозжечка в большое затылочное отверстие и ущемлению их на этом уровне.
Сущность данной аномалии заключается в гетеротопическом расположении мозжечка и продолговатого мозга в расширенном интраспинальном канале. Эта аномалия (мальформация) приводит к развитию сложных краниоспинальных синдромов, которые часто расцениваются неврологами как атипичная форма сирингомиелии и сирингобульбии, рассеянного склероза, а также опухолей задней черепной ямки или спинного мозга. Примерно у 80% пациентов аномалия Арнольда-Киари сочетается с патологией спинного мозга - сирингомиелией, которая характеризуется образованием в спинном мозге кист, вызывающих прогрессирующую миелопатию.
Данное заболевание названо в честь немецкого патологоанатома Юлиуса А. Арнольда (который описал в 1984 году данное заболевание) и австрийского патолога Ханса Киари (который также в 1895 году описал данную аномалию). Частота этого заболевания составляет от 3,3 до 8,2 наблюдений на 100 000 населения. Наиболее распространены аномалии Арнольда-Киари I и II типа; аномалии III и IV типов обычно несовместимы с жизнью:
• аномалия Арнольда-Киари I типа представляет собой опущение структур задней черепной ямки в позвоночный канал ниже плоскости большого затылочного отверстия;
• аномалия Арнольда-Киари II типа - происходит каудальная дислокация нижних отделов червя, продолговатого мозга и IV желудочка, нередко развивается гидроцефалия;
• аномалия Арнольда-Киари III типа встречается редко, характеризуется грубым каудальным смещением всех структур задней черепной ямки;
• аномалия Арнольда-Киари IV типа - гипоплазия мозжечка без смещения его вниз.
По данным Н.Н. Яхно и Д.Р. Штульмана (руководство для врачей «Болезни нервной системы» Москва, «Медицина», 2001): «… мальформация Киари представляет собой дисгенезию мозжечка в сочетании с широким кругом аномалий ромбовидного, среднего и промежуточного мозга. Выделяют несколько типов мальформации Киари.
Мальформация Киари I (взрослый тип) наиболее частая форма аномалий мозжечка. Этот симптомокомплекс представляет собой одно- или двустороннее опущение миндалин мозжечка через большое затылочное отверстие в позвоночный канал (миндалины - это нижняя часть мозжечка; в норме они расположены выше большого затылочного отверстия). Этому может способствовать каудальное смещение продолговатого мозга, нередко эктопия миндалин мозжечка сочетается с сирингомиелией и аномалиями краниовертебрального перехода. Важнейшим обстоятельством является тот факт, что клинические проявления возникают только на 3-4-м десятилетии жизни, не менее важно, что клинически асимптомная эктопия миндалин мозжечка не требует лечения, являясь случайной находкой на МРТ.
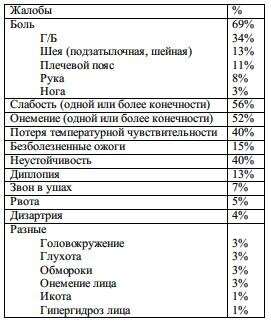
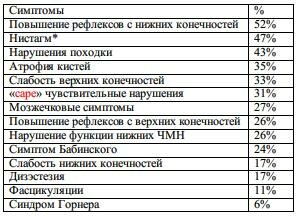
Частота симптомов при аномалии Киари I (по Paul и Jye, 1983 с изменениями): головная боль - 34%, боль в шее - 13%, опоясывающие боли в руках и/или ногах - 11%, слабость (как минимум в одной из конечностей) - 56%, онемение (как минимум в одной из конечностей) - 52%, потеря болевой и температурной чувствительности - 40%, пошатывание - 40%, горизонтальный и/или вертикальный нистагм - 30%, диплопия - 13%, дисфагия - 8%, рвота - 5%, дизартрия - 4%, головокружение - 3%, глухота - 3%, обмороки - 2%, онемение лица - 3%.
Как и при других процессах, поражающих цервико-медуллярный переход, при аномалии Киари I нередко наблюдается «нистагм, бьющий вниз». Наличие нарастающих очаговых симптомов (мозжечковых, стволовых, спинальных), а также гидроцефалии - повод к обсуждению показаний субокципитальной декомпрессии. В подобной ситуации необходим сугубо индивидуальный подход, чтобы избежать как неоправданного вторжения, так и промедления с хирургической коррекцией. Операция приводит к выздоровлению или улучшению у 2/3 больных. Благоприятным прогностическим признаком служит наличие только мозжечковых симптомов и дислокация мозжечка не ниже СI позвонка. Возможны рецидивы болезни на протяжении 3 лет после операции.
Мальформация Киари II (детский тип) складывается и смещения мозжечка, ствола и IV желудочка через большое затылочное отверстие. Характернейший признак - сочетание с менингомиелоцеле в поясничной области. Неврологические дефекты проявляются на фоне аномалий затылочной кости и шейного отдела позвоночника. Всегда имеется гидроцефалия, часто - стеноз водопровода мозга. Неврологические симптомы имеются уже при рождении. Менингомиелоцеле требует операции в первые дни жизни. Последующая субокципитальная декомпрессия может привести к значительному улучшению. Большинство больных нуждаются в шунтирующей операции, особенно если имеется стеноз водопровода мозга.
Мальформация Киари III. Менингоэнцефалоцеле в нижней затылочной или верхней шейной области в сочетании с пороками развития нижней части ствола мозга, основания черепа и верхних шейных позвонков. Мозговая грыжа включает мозжечок и в половине случаев - затылочную долю. Очень редкий симптомокомплекс с плохим прогнозом даже при своевременном хирургическом вмешательстве.
Мальформация Киари IV. аномалия представлена изолированной гипоплазией мозжечка и не является симптомокомплексом Киари в современном понимании.»
Порок Арнольда-Киари II типа
Киари 1
Продолжение
Термин «мальформация Киари» является предпочтительным по сравнению с традиционным «мальформация Арнольда-Киари» в связи со значительно большим вкладом, внесенным Киари.
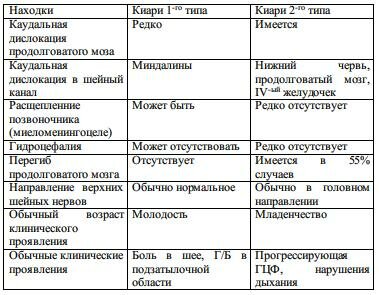


Мальформация Киари состоит из 4 типов аномалий заднего мозга, вероятно не связанных между собой. Большинство случаев приходится на типы 1 и 2 (см. табл. 6-5). На остальные варианты приходится крайне ограниченное количество случаев.
Табл. 6-5. Сравнительная характеристика 1-го и 2-го типов мальформации Киари (с изменениями)
Мальформация Киари 1-го типа
Т.н. первичная эктопия мозжечка. Это редкая аномалия, при которой имеется только каудальное смещение мозжечка с вклинением миндалин ниже БЗО (критерии см. МРТ ниже) и «удлинением миндалин типа затычки». В отличие от 2-го типа продолговатый мозг не смещен каудально (некоторые авторы с этим не согласны), ствол мозга не вовлечен, нижние черепные нервы не удлиненны, верхние шейные нервы не имеют направления в сторону головы.
Может быть фиброз мягкой и арахноидальной оболочек вокруг ствола мозга и миндалин. Могут быть гидромиелия и сирингомиелия СМ. Некоторые случаи могут быть приобретенными, после люмбо-перитонеального шунтирования или множественных (травматичных) ЛП.
Эпидемиология
Средний возраст клинического проявления 41 год (пределы: 12-73 года). Наблюдается некоторое преобладание & (&:%=1,3:1). Средняя продолжительность симптоматики определенно связанной с мальформацией Киари составляет 3,1 года (пределы: 1 мес – 20 лет). Если включить жалобы, которые носят неспецифический характер, напр., Г/Б, то этот срок составит 7,3 года. С наступлением эры МРТ эта задержка диагностики, возможно, станет меньше.
Клинические проявления
У пациентов с мальформацией Киари могут быть любые или даже все из следующих симптомов:
1. сдавление ствола мозга на уровне БЗО
2. ГЦФ
3. сирингомиелия
4. разделение внутричерепного отдела от спинального с периодическими повышениями ВЧД
Жалобы
Наиболее частой жалобой является боль (69%), особенно Г/Б, которая обычно ощущается в затылочной области (табл. 6-6).
Табл. 6-6. Жалобы при мальформации Киари 1-го типа (71 больной)
Г/Б часто вызывается разгибанием шеи и пробой Вальсальвы. Слабость также достаточно выражена, особенно при одностороннем пожимании руки. Может наблюдаться симптом Лермитта. Вовлечение нижних конечностей обычно проявляется двусторонней спастикой.
Симптомы
См. табл. 6-7. Три основных группы симптомов:
1. синдром сдавления в БЗО (22%): атаксия, кортико-спинальные и чувствительные нарушения, мозжечковые симптомы, паралич нижних ЧМН. 37% больных имеют тяжелые Г/Б
2. синдром центрального поражения СМ (65%): диссоциированные чувствительные нарушения (потеря болевой и температурной с сохраненной поверхностной и глубокой чувствительностью), иногда сегментарная слабость и признаки поражения длинных проводящих путей (сирингомиелический синдром). В 11% случаев наблюдается паралич нижних ЧМН
3. мозжечковый синдром (11%): атаксия туловища и конечностей, нистагм, дизартрия
Табл. 6-7. Симптомы при мальформации Киари 1-го типа (121 больной)
* в классическом случае: нистагм вниз при вертикальных движениях или ротаторный нистагм при горизонтальных движениях; также включает осциллопсию
Направленный вниз нистагм считается характерным для этого заболевания. В 10% случаев неврологический статус пациентов без изменений с единственной жалобой на затылочную Г/Б. В некоторых случаях основной жалобой может быть спастика.
Естественное течение
Естественное течение точно неизвестно (имеется только 2 публикации на эту тему). Пациент может оставаться в стабильном состоянии в течение нескольких лет с периодическими ухудшениями. В некоторых случаях возможно спонтанное улучшение (оспаривается).
Диагностика
Обзорные краниограммы
При анализе 70 обзорных краниограмм изменения были обнаружены только в 36% случаев (в 26% случаев была базиллярная импрессия, в 7% - платибазия; по одному пациенту имели болезнь Пэджета и вогнутый скат). Из 60 спондилограмм патологические изменения были в 35% случаев (включая ассимиляцию атланта, расширение канала, сращения шейных позвонков, агенезию задней дуги атланта).
МРТ
Диагностический метод выбора. На МРТ легко обнаружить большинство из выше указанных аномалий, включая вклинение миндалин, а также гидросирингомиелию, которая наблюдается в 20-30% случаев. Также видна вентральная компрессия ствола мозга, когда она имеется.
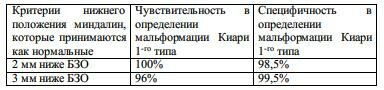
Положение миндалин мозжечка у 200 нормальных людей и 25 пациентов с мальформацией Киари 1-го типа приведено в табл. 6-8. Табл. 6-9 показывает изменения, которые возникают, если пределом нижнего положения миндалин считать 2, а не 3 мм.
Табл. 6-8. Расположение миндалин мозжечка от БЗО
* измерения относительно нижней части БЗО
Табл. 6-9. Критерии мальформации Киари 1-го типа
В отличие от выше приведенной информации в качестве нижней границы положения миндалин, пригодной для признания наличия мальформации Киари 1 го типа, часто указывают 5 мм.
Миелография
Ложно негативные результаты наблюдаются только в 6% случаев. КВ должно пройти вплоть до БЗО.
КТ
В связи с костными артефактами КТ имеет ограничения в визуализации области БЗО. Надежность исследования повышается при сочетании с интратекально введенным КВ (миелография). Находки: опущение миндалин и/или увеличение желудочков.
Лечение
Показания для хирургического лечения
Основываясь на том, что лучшие результаты наблюдаются в тех случаях, когда пациентов оперируют в течение первых 2 лет после появления симптомов, для симптоматических пациентов рекомендуется раннее хирургическое лечение. Асимптоматичных пациентов можно наблюдать и оперировать позднее, когда у них появятся симптомы. Пациентов, имевших симптомы, не менявшиеся в течение ряда лет, также можно наблюдать. Хирургическое лечение для них показано в случае ухудшения.
Хирургические методики
Наиболее частым видом операций является декомпрессия ЗЧЯ (подзатылочная краниоэктомия) с (или без) другими вмешательствами (обычно комбинируется с пластикой ТМО и шейной ламинэктомией С1 до С2 или С3.
Пациента кладут на живот с валиками под грудной клеткой. Голову фиксируют головодержателем Майфилда. Шею сгибают для увеличения промежутка между затылочной костью и задней дужкой С1. Плечи отводят вниз с помощью липкой ленты. Одно бедро должно быть уложено на валик (мешочек с песком) на случай, если понадобится взять фрагмент широкой фасции для пластики. Разрез делают по средней линии от иниона до ∼ остистого отростка С5. Резецируют кость выше БЗО на ∼3 см вверх и ∼3 см в ширину. ТМО вскрывают Y-образным разрезом и иссекают верхний лоскут. ВНИМАНИЕ: при мальформации Киари поперечные синусы располагаются обычно очень низко (поэтому резекция затылочной кости должна быть небольшой, основной упор должен быть сделан на шейную ламинэктомию и резекцию краев БЗО для того, чтобы устранить компрессию миндалин; компрессия миндалин происходит не в ЗЧЯ). Кроме того, избыточная резекция затылочной кости может привести к выбуханию полушарий мозжечка в образовавшийся дефект, что чревато дополнительными проблемами.
Произведите пластику ТМО надкостницей или фрагментом широкой фасции бедра, чтобы обеспечить достаточно места для миндалин и продолговатого мозга. Некоторые хирурги также дополняют операцию тампонадой верхушки IV-го желудочка мышцей или тефлоном, дренированием сирингомиелитической кисты, если она имеется (фенестрация, обычно через место входа задних корешков, с/или без установкой стента или шунта), шунтированием IV-го желудочка, терминальной вентрикулостомией, вскрытием отверстия Мажанди, если оно закрыто).
Некоторые авторы настоятельно советуют не пытаться удалять спайки между миндалинами, чтобы избежать случайного повреждения жизненно важных структур, включая ЗНМА. Другие же рекомендуют осторожное разделение миндалин и даже их обработку биполярной коагуляцией с тем, чтобы уменьшить их объем.
Некоторые авторы рекомендуют производить трансоральную резекцию ската и зубовидного отростка в случае наличия вентральной компрессии ствола мозга, т.к. они считают, что у этих пациентов может наступить ухудшение после того, как будет произведена только декомпрессия ЗЧЯ. В связи с тем, что было показано, что возникающие при этом изменения являются обратимыми, представляется более целесообразным произвести это вмешательство в случае наступления ухудшения или прогрессирования базиллярной импрессии на МРТ после декомпрессии ЗЧЯ.
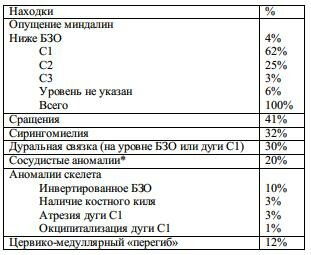
Оперативные находки
См. табл. 6-10. Вклинение миндалин имеется во всех случаях (по определению); наиболее часто они расположены на уровне С1 (62%). В 41% случаев имеются сращения между ТМО, арахноидальной оболочкой и миндалинами с закупоркой отверстий Люшка и Мажанди. В 40% случаев отделение миндалин происходит легко.
Табл. 6-10. Оперативные находки при мальформации Киари 1-го типа (71 пациент)
* сосудистые аномалии: ЗНМА была расширена или имела необычное расположение у 8 пациентов (ЗНМА часто спускается до нижнего края миндалин); большие дуральные венозные лакуны
Хирургические осложнения
После подзатылочной краниоэктомии плюс ламинэктомии С1-3 у 71 пациента (с пластикой ТМО у 69 из них) у одного пациента через 36 ч после операции наступила смерть в результате апноэ во время сна. Наиболее частым послеоперационным осложнением было угнетение дыхания (у 10 пациентов), обычно в течение 5 дней, чаще ночью. В связи с этим рекомендуется тщательное наблюдение за дыханием. Другие риски операции: ликворея, вклинение полушарий мозжечка, сосудистые повреждения (ЗНМА и т.д.).
Результаты операций
Пациенты с жалобами на боль обычно хорошо реагируют на операцию. Предоперационная слабость обычно хуже поддается лечению, особенно если уже имеется атрофия мышц. Чувствительность может улучшиться, если задние столбы не поражены, и дефицит обусловлен вовлечением только спинно-таламических трактов.
Ротон считает, что основным эффектом операции является прекращение прогрессирования симптоматики.
Наиболее благоприятные результаты наблюдались у пациентов с мозжечковым синдромом (улучшение у 87% без последующего ухудшения). Факторы, которые коррелировали с плохими исходами: наличие атрофии, атаксия, сколиоз, продолжительность симптоматики более 2 лет.
Табл. 6-11. Отдаленные результаты (69 пациентов, средний срок наблюдения 4 года)
* у этих пациентов было ухудшение до предоперационного уровня (без дальнейшего ухудшения) в течение 2-3 лет после операции; рецидив наступил у 30% пациентов с синдромом компрессии в БЗО и у 21% пациентов с синдромом центрального поражения СМ
Мальформация Киари 2-го типа
Обычно сочетается с миеломенингоцеле (ММЦ) и реже с закрытым расщеплением дужек позвоноков (spina bifida ocсulta).
Патофизиология
Вероятно не связана с фиксацией СМ сопутствующим ММЦ. Более вероятна первичная дисгенезия ствола мозга со множеством других аномалий развития.
Основные находки
Каудальная дислокация цервико-медуллярного сочленения, моста, IV-го желудочка и продолговатого мозга. Миндалины мозжечка расположены на уровне БЗО или ниже его. Вместо обычного изгиба цервико-медуллярного сочленения имеется его деформация в виде перегиба.
Другие возможные находки:
1. клювовидный изгиб четверохолмной пластинки
2. отсутствие прозрачной перегородки с увеличенной межталамической спайкой: считается, что отсутствие прозрачной перегородки обусловлено некрозом с резорбцией в результате ГЦФ, а не врожденного отсутствия
3. слабо миелинизированные мозжечковые листи
4. ГЦФ: имеется в большинстве случаев
5. гетеротопия (патологическая дислокация)
6. гипоплазия фалькса
7. микрогирия
8. дегенерация ядер нижних ЧМН
9. костные аномалии:
A. в области цервико-медуллярного сочленения
B. ассимиляция атланта
C. платибазия
D. базиллярная импрессия
E. деформация Клиппеля-Фейля
10. гидромиелия
11. образование лакун в черепе
Клинические проявления
Проявления связаны с дисфункцией ствола мозга и нижних ЧМН. Манифестация заболевания во взрослом возрасте наблюдается редко. Проявления у новорожденных существенно отличаются от проявлений у детей старшего возраста. Новорожденные более склонны к развитию быстрого неврологического ухудшения со значительной дисфункцией ствола мозга в течение нескольких дней. У более старших детей симптомы развиваются более постепенно и редко проявляются в столь тяжелом виде.
Находки:
1. затруднения глотания (нейрогенная дисфагия) (69%). Проявляется плохим кормлением, цианозом после кормления, назальной регургитацией, продолжительным временем кормления, скоплением слюны. Глоточный рефлекс часто понижен. Более тяжело выражены у новорожденных
2. припадки апноэ (58%): связаны с нарушенной стимуляцией дыхания. Чаще встречается у новорожденных
3. стридор (56%): чаще у новорожденных, обычно хуже на вдохе (при ларингоскопии виден паралич отводящих и иногда приводящих голосовых связок) в связи с парезом Х-го ЧМН. Обычно носит временный характер, но может перейти в остановку дыхания
4. аспирация (40%)
5. слабость верхних конечностей (27%), которая может перейти в тетрапарез
6. опистотонус (18%)
7. нистагм: особенно направленный вниз
8. слабый крик или отсутствие его
9. слабость лицевой мускулатуры
Диагностика
Обзорные краниограммы
Может быть видна цефалофациальная диспропорция в результате врожденной ГЦФ. Образование лакун в черепе (т.н. lukenschadel) наблюдается в 85% случаев (округлые дефекты в черепе с четкими краями, разделенные неравномерно ветвящимися костными полосками). Могут быть низко расположенный внутренний затылочный выступ (укороченная ЗЧЯ), увеличение БЗО (в 70%) и удлинение верхнешейных дужек.
КТ и/или МРТ
• основные находки
A. Z-образная деформация продолговатого мозга*
B. мозжечковая «затычка»
C. четверохолмное сращение (клювовидный изгиб в области четверохолмия)
D. увеличенная промежуточная масса (межталамическое сращение)*
E. удлинение/цервикализация продолговатого мозга
F. низкое прикрепление мозжечкового намета
• сопутствующие находки
A. ГЦФ
B. сирингомиелия в области цервико-медуллярного сочленения (частота по данным до использования МРТ составляет 48-88%)
C. отключенный IV-ый желудочек
D. церебелло-медуллярная компрессия
E. агенезия/дисгенезия мозолистого тела*
* эти признаки лучше оценивать по МРТ
Ларингоскопия
Ее производят у детей со стридором для исключения крупа и других инфекций верхних дыхательных путей.
Лечение
• установите шунт для коррекции ГЦФ (если шунт уже имеется, то проверьте его функционирование)
• при нейрогенной дисфагии, стридоре, приступах апноэ рекомендуется ускоренная декомпрессия ЗЧЯ (требуется у 18,7% пациентов с ММЦ); перед проведением рекомендованной декомпрессии всегда необходимо убедиться в том, что у пациента имеется функционирующий шунт!
Хирургическая декомпрессия
NB: плохие оперативные результаты у новорожденных объясняются тем, что многие из неврологических находок могут быть связаны с врожденными (некорректиуемыми) нарушениями, которые не может улучшить декомпрессия. По другой точке зрения, гистологические изменения обусловлены хронической компрессией ствола мозга и сопутствующей ишемией, поэтому ускоренная декомпрессия ствола мозга должна быть произведена как только появляются какие-либо из следующих критических предупреждающих симптомов: нейрогенная дисфагия, стридор, приступы апноэ.
Методика операции: декомпрессия миндалин мозжечка обычно с пластикой ТМО. Пациента укладывают на живот, шея согнута. Производят подзатылочную краниоэктомию и шейную ламинэктомию, которая должна достигать вершины миндалин мозжечка. Между краем БЗО и дужкой С1 обычно удается обнаружить утолщенную дуральную связку, сдавливающую СМ. ТМО вскрывают Y-образным разрезом. При вскрытии ТМО выше уровня БЗО у младенцев требуется осторожность, т.к. они имеют хорошо развитый затылочный синус и могут иметь большие дуральные «озера». Не пытайтесь отделить миндалины от подлежащего продолговатого мозга. В том случае, когда имеется большая сирингомиелитическая полость, делают сиринго-субарахноидальное шунтирование.
Если до операции имеется стридорозное дыхание или парез мышц, отводящих гортань, рекомендуется трахеостомия (обычно временная). Требуется тщательное послеоперационное наблюдение за дыханием (контроль обструкции и снижения стимуляции вентиляции). ИВЛ показана при гипоксии и гиперкарбии.
Исходы
В 68% случаев наступает полное или практически полное разрешение симптомов. В 12% случаев имеется незначительный или умеренный остаточный неврологический дефицит. В 20% случаев улучшения не наступает. В общем, у новорожденных результаты хуже, чем у детей старшего возраста.
Наиболее частой причиной смертности является остановка дыхания (8 из 17 умерших пациентов). Остальные случаи: менингит/вентрикулит (6 пациентов), аспирация (2) и билиарная атрезия (1).
При послеоперационном наблюдении продолжительностью от 7 мес до 6 лет смертность оперированных больных составила 37,8%.
Наиболее важными прогностическими факторами были предоперационное состояние и скорость неврологического ухудшения. Летальность среди младенцев с кардио-пульмональной остановкой, параличом голосовых связок и слабостью рук в течение 2 нед после клинического проявления составила 71%. При более постепенном ухудшении летальность составила 23%. Наихудшим прогностическим фактором в отношении результативности оперативного лечения был двусторонний паралич голосовых связок.
Другие типы мальформации Киари
Мальформация Киари 3-го типа
Встречается редко. Является наиболее тяжелой формой. Смещение структур ЗЧЯ с вклинением мозжечка через БЗО в шейный канал, часто с шейным или подзатылочным энцефаломенингоцеле. Обычно не совместимо с жизнью.
Мальформация Киари 4-го типа
Гипоплазия мозжечка без вклинения мозжечка.
Гринберг. Нейрохирургия
Латышева В.Я., Олизарович М.В., Филюстин А.Е., Гурко Н.А., Учреждение образования «Гомельский государственный медицинский университет», Республиканский научно-практический центр радиационной медицины и экологии человека, г. Гомель, Республика Беларусь (7 (45) 2011 / Оригинальные исследования /Original Researches/).
Заболевание названо в честь австрийского патолого-анатома Ханса Киари [Hans Chiari], который в 1891 году описал несколько типов аномалий развития ствола мозга и мозжечка.
Краниовертебральные аномалии - это врожденные или приобретенные дефекты развития структур головного и спинного мозга краниовертебрального перехода или костных структур основания черепа и двух верхних шейных позвонков (платибазия, базилярная импрессия, атлантоаксиальный подвывих, ассимиляция атланта), которые служат основанием диагностики первичного или вторичного генеза клинических проявлений. Одним из проявлением Краниовертебральной аномалии является аномалия, или порок, или мальформация Киарая может проявляться синдром Арнольда-Киари (I, II, III). Мальформация Арнольда- Киари - врожденное нарушение строения головного мозга, которое характеризуется низким расположением миндалин мозжечка, т.е. это церебелло-медуллярное уродство в виде врожденного опущения продолговатого мозга и миндалин мозжечка в суженное большое затылочное отверстие. Помимо врожденного (первичного) происхождения) низкого расположение миндалин мозжечка последнее может быть также и приобретенным (вторичным) в результате частых люмбальных пункций или после люмбоперитонеального шунтирования.
Мальформация Арнольда-Киари характеризуется внутричерепной гипертензией, стволовыми нарушениями, каудальной дислокацией миндалин мозжечка и платибазией, при которой происходит вдавление основания затылочной кости и ската в заднюю черепную ямку. Платибазия - это уплощение основания черепа, в результате чего скат расположен более горизонтально по отношению к плоскости передней черепной ямки. Платибазия клинически не проявляется, но в сочетании с другой костной аномалией черепа может вызывать различные симптомы. Сдавление спинного мозга приводит к двигательным нарушениям (спастическим парапарезам), а ствола мозга - к вовлечению в патологический процесс V–XII пар черепных нервов и нередко сопровождается зрительными нарушениями. Современная патоморфология выделяет три основных типа этой аномалии (мальформации).
Таким образом при Ааномалии Арнольда-Киари I - II клинических проявления заболевания объясняются затруднением оттока ликвора из полости черепа в спинальное субарахноидальное пространство вследствие сужения большой затылочной цистерны при опускании миндалин мозжечка. Это приводит к повышению интракраниального и снижению интраспинального ликворного давления. Самым частым симптомом является головная боль. Особенно характерна боль в затылочной области, усиливающаяся при покашливании и натуживании, боли в шее, слабость и нарушение чувствительности рук, неустойчивая походка, диплопия, смазанная речь, затрудненное глотание, рвота и шум в ушах. При аномалии Киари низко расположенные миндалины мозжечка затрудняют свободную циркуляцию спинномозговой жидкости между головным и спинным мозгом. Миндалины блокируют большое затылочное отверстие, в результате чего нарушается отток ликвора и развивается гидроцефалия.



Интсрументальная диагностика. Патология краниоспинального перехода чаще выявляется при компьютерной или магнитно-резонансной томографии (КТ, МРТ). Наиболее распространены аномалии Арнольда- Киари I и II типа. Патогномоничным признаком аномалии является опускание миндалин мозжечка более чем на 3–5 мм ниже плоскости большого затылочного отверстия. Наряду с общепринятыми методиками выявления данной патологии при лучевой диагностике и МРТ-исследовании у взрослых возможна ранняя ультразвуковая диагностика мальформации у плода в сроках гестации от 22 до 28 недель. Используется горизонтальная и фронтальная плоскости сканирования, при этом выявляемость признаков мальформации Киари составляет от 24 до 45 %.
Результаты исследования пациентов с аномалией Арнольда-Киари I - II свидетельствуют о том, что наиболее частыми жалобами у них являются головная боль и головокружение, часто в сочетании, общая слабость(как по отдельности, так в сорчетаии). Несколько реже пациенты жалуются на боль в шейном отделе позвоночника, снижение настроения, парестезии в руках и ногах, пошатывание при ходьбе. В ряде случаев данные, полученные при неврологическом обследовании пациентов, позволяют установить топический диагноз до выполнения томографического исследования. Наиболее частыми неврологическими симптомами при синдроме Арнольда-Киари являются мозжечковая атаксия, когнитивное снижение и патологический симптом Бабинского с двух сторон. В некоторых случаях при пролабировании миндалин мозжечка отмечается только головная боль без неврологического дефицита или возможно легкое пошатывание при ходьбе и тремором рук. Наиболее выраженные затруднения при проведении дифференциальной диагностики («демиелинизирующее заболевание центральной нервной системы. Подозрение на рассеянный склероз» вызывают пошатывание при ходьбе, изменение походки у лиц молодого возраста, но проведение МРТ-исследования позволяет исключить демиелинизирующее заболевание центральной нервной системы. При анализе томограмм головного мозга выявляется следующая патология, с которой чаще всего сочетается аномалия Арнольда-Киари: субатрофия полушарий головного мозга, хронические воспалительные процессы околоносовых пазух, кисты околоносовых пазух, менингомиелоцеле, арахноидальная киста турецкого седла, кистозно-глиозные изменения после удаления опухоли больших полушарий, патологическая извитость вертебральной артерии в интракраниальном сегменте,,артериовенозная мальформация полушария мозжечка, ретроцеребеллярная киста, синдром Штурге-Вебера, опухоль мосто-мозжечкового угла, гидроцефалия, липома мозолистого тела, опухоль мозжечка.
Дифференциальная диагностика синдрома Арольда-Киари с такими проявлениями как головные боли, атаксия, головокружение, тетрапарез, бульбарные нарушения чаще всего проводили с опухолью задней черепной ямки или краниоспинального перехода, рассеянным склерозом; рефлекторными, корешковыми синдромами; синдромом заднего шейного симпатического узла при шейном остеохондрозе; сосудистыми нарушения ствола мозга при вертебробазилярной недостаточности; патологическими изменениями основаниячерепа (платибазия). Представленный перечень свидетельствует о клинической значимости врожденной аномалии, которая может манифестировать различными заболеваниями и затруднять выбор патогенетической терапии.
http://www.pediatricneurosciences.com/article.asp?issn=1817-1745;year=2011;volume=6;issue=2;spage=116;epage=117;aulast=Ambekar
http://www.pediatricneurosciences.com/article.asp?issn=1817-1745;year=2008;volume=3;issue=2;spage=169;epage=171;aulast=Garg
http://www.medscape.com/content/2004/00/47/06/470602/470602_fig.html
Мальформация Арнольда-Киари
ID: 2592 Chiari 1 malformation Dr Frank Gaillard - 7 May 2008 Incidental finding
ID: 10769 Chiari 1 malformation measurement Dr Frank Gaillard - 19 Sep 2010 The vertical distance from the tip of the cerebellar tonsils to a line d...
ID: 15819 Chiari 1 malformation with focal syrinx Dr Frank Gaillard - 14 Nov 2011 This case illustrates a Chiari 1 malformation complicated by a focal syr...
ID: 22950 Chiari 1 malformation with syrinx Dr Peter Mitchell - 7 May 2013 This case illustrates how a syrinx can be distal to the posterior fossa ...
ID: 10358 Chiari 1 malformation, syrinx and scoliosis Dr Frank Gaillard - 8 Aug 2010 Child with a Chiari 1 malformation and resulting extensive syrinx with r...
ID: 13619 Chiari I & sinus pericranii Dr Hani Alsalam - 27 Apr 2011 Chiari I and sinus pericranii.
ID: 5993 Chiari I and severe copper beaten skull Dr Frank Gaillard - 4 Apr 2009 This 6 month old child has muliple sutural synostosis (sagittal suture a...
ID: 21023 Chiari I malformation Dr Bita Abbasi - 29 Dec 2012
ID: 3609 Chiari I malformation Dr Frank Gaillard - 28 May 2008 This MRI performed for another indication demonstrates low lying cerebel...
ID: 12712 Chiari I malformation Dr Gagandeep Choudhary - 2 Jan 2011 MRI demonstrates low lying cerebellar tonsills, embracing the medulla, c...
ID: 16632 Chiari I malformation Dr Miguel Terrazas Terrazas - 5 Feb 2012
ID: 18617 Chiari I malformation : CT morphology Dr Charlie Chia-Tsong Hsu - 15 Jul 2012
ID: 24077 Chiari I malformation and extensive cervico-dorsal syringohydromyelia Dr Mohammed A ElBeialy - 26 Jul 2013
ID: 18924 Chiari I malformation with dorso-lumbar syrinx Dr Maxime St-Amant - 27 Jul 2012 Related articles : Chiari malformation Chiari I malformation syrinx &am...
ID: 8385 Chiari I malformation with post-op pseudomeningocoele Dr Frank Gaillard - 27 Jan 2010 Chiari I malformation with posterior decompression and pseudomeningocoele.
ID: 16995 Chiari I malformation with syringomyelia Dr Praveen Jha - 7 Mar 2012
ID: 5686 Chiari I malformation with syrinx Dr Frank Gaillard - 14 Mar 2009 Chiari I malformation with large syrinx.
ID: 16050 Chiari II malformation Dr Frank Gaillard - 11 Dec 2011 This case illustrates many of the common features of Chiari II malformat...
ID: 5420 Chiari II malformation Dr Frank Gaillard - 19 Jan 2009 This patient also has spina bifeda, and the features are consistent with...
ID: 16044 Chiari II malformation Dr Frank Gaillard - 10 Dec 2011
ID: 17054 Chiari II malformation Dr Roberto Schubert - 13 Mar 2012 Findings consistent with Chiari type II malformation.
ID: 16048 Chiari II malformation Dr Frank Gaillard - 11 Dec 2011 This patient was born with spina bifeda and a myelomeningocoele which wa...
ID: 16049 Chiari II malformation Dr Frank Gaillard - 11 Dec 2011 This patient was born with a sacral myelomeningocoele which was reparied...
ID: 23550 Chiari II malformation with spinal meningomyelocele Dr Mohammed A ElBeialy - 23 Jun 2013 Chiari II malformation with large spinal dysraphism, spinal myelomeningo...
ID: 18557 Chiari II malformation: CT morphology Dr Charlie Chia-Tsong Hsu - 12 Jul 2012
ID: 16055 Chiari III malformation - repaired Dr Frank Gaillard - 12 Dec 2011 This case highlights the difficulty in assessing congential malformation...
ID: 18005 Chiari malformation - type 1 + syrinx Dr Miguel Terrazas Terrazas - 31 May 2012
ID: 16113 Chiari type I malformation Dr Franco Ruales - 20 Dec 2011
ID: 24527 Chiari type II malformation Dr Ahmed Abd Rabou - 23 Aug 2013 The patient had lumbar meningocele operated shortly after birth with pla...
ID: 24245 Chiari type II malformation Dr Ahmed Abd Rabou - 3 Aug 2013
ID: 24246 Chiari type II malformation - on ultrasound Dr Ahmed Abd Rabou - 3 Aug 2013
ID: 6648 Chiari type III malformation Dr Paresh K Desai - 28 Jul 2009 3 year old child with occipital swelling.
Из литературных источников