Болезнь Канавана-ван Богарта-Бертранда также относится к лейкодистрофиям. Предполагается, что процесс распада миелиновой оболочки начинается еще в период внутриутробной жизни ребенка. Первые симптомы заболевания в большинстве случаев обнаруживаются уже с момента рождения. Отмечается сонливость, ребенок мало двигается, снижен аппетит. В ряде случаев болезнь может начинаться с появления судорог. Затем в возрасте 2-6 месяцев отмечается снижение тонуса мышц шеи, повышение мышечного тонуса в конечностях, иногда хорееформные гиперкинезы, атрофия сосков зрительных нервов, косоглазие, непроизвольные движения глазных яблок, увеличение размеров головы вследствие скопления большого количества жидкости (гидроцефалия). Прикосновение к ребенку, сильный шум вызывают состояние опистотонуса. Быстро утрачиваются слух, зрение, наступает слабоумие. В конечной стадии заболевания больные находятся в состоянии децеребрации, обездвижены, выражены расстройства кровообращения и дыхания. Смерть наступает в возрасте от шести месяцев до двух лет.
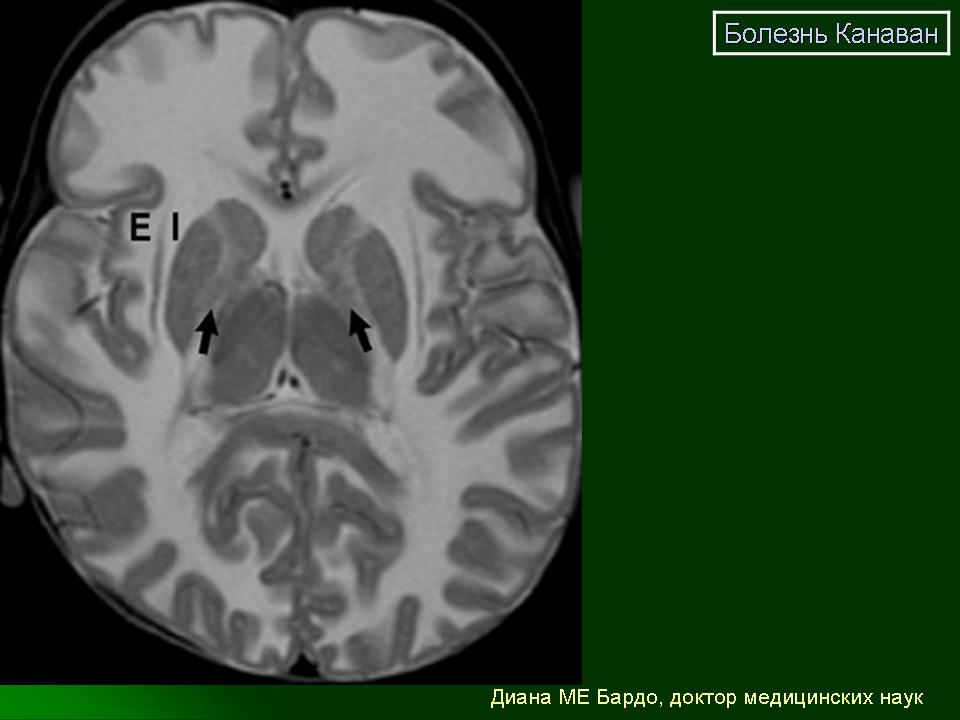
Случай лейкодистрофии Канавана-Ван Богарта-Бертранда.
С. В. Михайлова Е. Ю. Захарова А. М. Букина Е. С. Ильина А. Я. Покровская И. Д. Федонюк Р. Ц. Бембеева А. С. Петрухин
Лейкодистрофии - довольно редкая группа наследственно-дегенеративных заболеваний нервной системы, обусловленных дефектами различных белков, участвующих в формировании миелина. Термин «лейкодистрофия» был предложен Bielshowsky и Henneberg в 1928 г. [1, 3].
В настоящее время известно несколько форм лейкодистрофий, при которых первичный дефект установлен на биохимическом и/или молекулярно-генетическом уровне: метахроматическая лейкодистрофия, болезнь Краббе, болезнь Канавана (БК), Х-сцепленная адренолейкодистрофия, болезнь Александера, болезнь Пелицеуса - Мерцбахера. Для ряда наследственных заболеваний из группы лейкодистрофий, первичный дефект пока не выявлен - это мегалэнцефалия и лейкодистрофия (болезнь Ван дер Кнаапа, или вакуолизирующая лейкодистрофия с субкортикальными кистами), синдром Коккейна, синдром Айкарди - Гутерини.
В литературе встречаются разные названия лейкодистрофии Канавана - Ван Богарта - Бертранда (МIM 271900): спонгиозная дегенерация мозга, инфантильная спонгиоформная дегенерация ЦНС, инфантильная спонгиозная дегенерация, аспартоацилазная недостаточность, N-ацетиласпартиковая ацидурия [4, 29].
Первое клиническое описание основных клинических симптомов БК принадлежит Globus и Strauss (1928 г.) [29]. В 1931 г. Канаван сообщил о ребенке с прогрессирующим ростом головы, у которого при аутопсии была выявлена спонгиозная дегенерация белого вещества головного мозга; заболевание было определено как диффузный периаксиальный энцефалит Шильдера [29]. Позднее была выдвинута гипотеза о семейном характере заболевания [29]. В 1949 г. Ван Богарт и Бертранд определили спонгиозную дегенерацию мозга у 3 детей еврейской национальности. С тех пор описаны многочисленные случаи этого заболевания [10, 15, 21, 28, 32].
Биохимический дефект при БК был идентифицирован
R. Matalon и соавт. в 1988 г. [22]. Они обнаружили повышение уровня N-ацетиласпартата в моче и недостаточность аспартоацилазы в культуре клеток фибробластов у 3 пациентов с БК [22].
В 1993 г. клонирован ген аспартоацилазы (ASPA) и обнаружены мутации, приводящие к БК [20, 24].
Эпидемиология, этиология и генетика
БК наследуется по аутосомно-рецессивному типу. Частота заболевания в общей популяции не установлена ввиду его редкости. Болезнь встречается во всех этнических группах, но с наибольшей частотой среди евреев ашкенази (1 случай на 5000 новорожденных) [29].
Причиной БК являются мутации гена, кодирующего фермент аспартоацилазу. Ген картирован на 17-й хромосоме. В настоящее время в нем описано около 50 различных мутаций, в подавляющем большинстве точковые. По данным
R. Matalon и соавт. [23], среди евреев ашкенази наиболее часто встречаются мутации Е285А (82,9% аллелей) и Y231X (14,8% аллелей). Среди европейцев нееврейского происхождения частой является мутация А305Е (60% аллелей) [5].
В России частота и спектр мутаций, определяющих развитие заболевания, не изучены, и сообщений о случаях БК в доступной литературе мы не нашли.
Биохимия и патогенез
БК, как уже говорилось, обусловлена недостаточностью фермента аспартоацилазы, что приводит к накоплению N-ацетиласпартата в мозге, спинномозговой жидкости, плазме крови и моче [22]. Аспартоацилаза катализирует расщепление N-ацетил-L-аспартата на L-аспартат и ацетат [4, 18, 19, 30].
N-Aцетиласпартат у человека был обнаружен в больших концентрациях только в клетках нервной ткани. Насколько велика его концентрация, настолько мало известно о его роли в организме.
Согласно одной из гипотез, N-ацетиласпартат за счет высокой осмолярности представляет компонент «водного насоса» в головном мозге, а фермент аспартоацилаза участвует в гидролизе N-ацетиласпартата, регулируя содержание воды в межклеточном пространстве [18, 29].
Молекулярные механизмы патогенеза БК неясны, так как мало известно о функции N-ацетиласпартата. Остается также невыясненным, почему первые симптомы заболевания появляются в первые месяцы жизни, хотя повышение концентрации N-ацетиласпартата выявляется еще пренатально [29, 30].
На сегодняшний день достаточно ясно, что вряд ли основной причиной клинических симптомов заболевания является недостаточность продуктов блокированной реакции - ведь существует много других источников и аспартата, и ацетата в мозге [4, 29]. Предполагается токсическое действие N-ацетиласпартата или его метаболитов на ткань мозга, что, по всей вероятности, приводит к возникновению хронического отека.
Основные патоморфологические находки локализуются в белом веществе, которые замещаются развитой сетью кистозных полостей. При морфологическом исследовании выявляется губчатая (спонгиозная) вакуолизация в нижних слоях серого вещества, в субкортикальных отделах белого вещества мозга и мозжечка. При электронной микроскопии выявляются вакуоли в «набухшей» цитоплазме и отростках протоплазматических астроцитов в коре, а также вакуолизация белого вещества и аномальные митохондрии в астроцитах [6, 7, 16].
Данные лабораторных и функциональных исследований
У пациентов с БК при КТ и МРТ головного мозга обнаруживают диффузную дегенерацию белого вещества с вовлечением в патологический процесс полушарий мозга, в меньшей степени поражаются мозжечок и ствол мозга [31]. В ранних стадиях патологического процесса при КТ и МРТ патология может не выявляться [11, 26]. Однако на начальных этапах БК при МРТ может определяться поражение белого вещества в перивентрикулярной области, которое часто расценивают как перивентрикулярную лейкомаляцию вследствие перинатального поражения ЦНС гипокcически-ишемического генеза. По мере прогрессирования заболевания при МРТ отмечается диффузная дегенерация белого вещества в виде очагов повышенного сигнала в белом веществе в Т2-режиме [27].
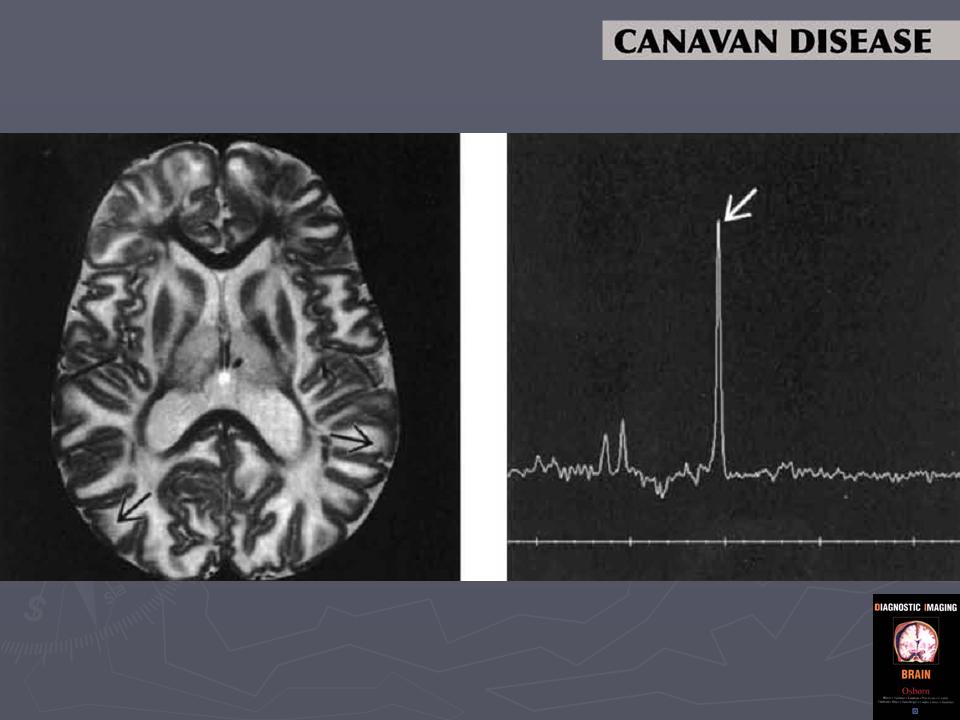
Как показано в исследованиях K. Gripp и соавт. [14], отличие от других форм лейкодистрофий и демиелинизирующих заболеваний при магнитно-резонансной протонной спектроскопии у пациентов с БК отмечалось резкое повышение и уровня N-ацетиласпартата, и соотношения N-ацетиласпартат/креатинин, что может служить важным критерием для верификации диагноза [9, 14]. При офтальмологическом исследовании выявляют атрофию дисков зрительных нервов [27].
Определение концентрации N-ацетиласпартата в моче и измерение активности аспартоацилазы в культуре кожных фибробластов являются надежными биохимическими тестами при установлении диагноза БК [23]. Уровень N-ацетиласпартата в моче, плазме, спинномозговой жидкости определяют хроматографическими методами, такими как хроматомасс-спектрометрия и высокоэффективная жидкостная хроматография (ВЭЖХ).
Профилактика
Пренатальная диагностика БК в отягощенных семьях проводится путем определения концентрации N-ацетиласпартата в амниотической жидкости или методами ДНК-анализа ворсин хориона, если генотип пробанда установлен [29].
В Российской детской клинической больнице (РДКБ) нами наблюдался пациент с БК в возрасте 5,5 лет. Было проведено тщательное клиническое обследование, включавшее клинический осмотр в динамике, клинические и биохимические анализы крови и мочи по стандартным методикам.
Из инструментальных методов использовали офтальмоскопию, ультразвуковое исследование внутренних органов. Неврологическое обследование проводилось с применением компьютерной электроэнцефалографии, КТ и МРТ головного мозга.
Спектр и концентрацию органических кислот в моче определяли методом ВЭЖХ [2, 23]. Для выявления распространенных мутаций в гене аспартоацилазы использовали стандартные методы полимеразной цепной реакции с последующим рестрикционным анализом и прямым нерадиоактивным секвенированием [23, 24, 30].
Приводим это наблюдение.
Больной Б., 5,5 лет. Наблюдается в РДКБ на протяжении 4 лет. Впервые поступил в возрасте 2 лет с жалобами на задержку психического и двигательного развития, снижение остроты зрения, генерализованные тонико-клонические судороги, увеличение окружности головы.
Из анамнеза: ребенок от молодых здоровых родителей (матери 22 года, отцу 23 года, русские), от первой беременности, протекавшей физиологически. Роды в срок с родостимуляцией, масса тела при рождении 3950 г, длина 54 см, окружность головы 36 см (98 центилей).
С рождения отмечались беспокойство, нарушение сна, частые срыгивания, симптом Грефе, напряжение и пульсация большого родничка, мышечный гипертонус, преимущественно в дистальных отделах конечностей. Проводилась дегидратирующая, седативная терапия.
К 3-му месяцу жизни ребенок самостоятельно не удерживал голову, непостоянно прослеживал глазами за предметами, отмечались крупноразмашистый спонтанный и провоцируемый тремор, усиление всех безусловных рефлексов.
В 4 мec: окружность головы 47 см (99,6 центили), практически полное отсутствие эфферентных реакций, малоэмоциональность, вялость зрачковых реакций, снижение слуха, псевдобульбарный синдром, патологические синкинезии, «стартл-рефлексы» и отсутствие формирования выпрямляющих установочных реакций.
В 5 мес находился на стационарном лечении с диагнозом: последствия перинатального поражения ЦНС, спастический тетрапарез, субкомпенсированная гидроцефалия. Задержка психического и физического развития, нейросенсорная тугоухость справа II степени. Частичная атрофия зрительных нервов. Функциональная кардиопатия (открытое овальное окно, дополнительная трабекула в полости левого желудочка).
В 8 мес по сравнению с предыдущими осмотрами неврологический статус оставался прежним, однако отмечалось нарастание мышечного тонуса. В 1,5 года на фоне гипертермии впервые появились генерализованные тонико-клонические судороги. Ребенок неоднократно поступал в реанимационное отделение РДКБ по поводу нарастания дыхательной недостаточности центрального и инфекционного генеза, что сопровождалось ухудшением неврологического статуса: угнетение сознания до сопора, учащение судорог и эпизодов апноэ, нарастание мышечного тонуса до выраженного гипертонуса с последующим частичным восстановлением функций при купировании инфекционного процесса. За время наблюдения перенес деструктивную пневмонию правого легкого, осложнившуюся абсцессом верхней доли. Проводилась курсами симптоматическая терапия (вазоактивная, ноотропная, антиэпилептическая, витамины группы В и др.) без явного положительного эффекта: сохранялись грубое нарушение психомоторного развития, редкие судороги, снижение зрения, тетрапарез спастического характера, псевдобульбарный паралич.
Данные объективного исследования
При поступлении в отделение в возрасте 5,5 лет: состояние тяжелое. Соматический статус: кожные покровы смуглые, с бронзовым оттенком, слизистые оболочки розовые. Дыхание жесткое, хрипов нет. Границы сердца соответствуют возрастной норме. На верхушке сердца во втором - третьем межреберье слева выслушивается систолический шум. Живот мягкий, безболезненный, доступен глубокой пальпации. Печень и селезенка не увеличены. Дизурических явлений нет.
Неврологический статус: общемозговых и менингеальных симптомов нет. Форма головы - гидроцефальная с выраженными лобно-теменными буграми. Окружность головы 57 см, перкуторный звук коробочный. Большой родничок закрыт. Множественные стигмы дизэмбриогенеза: гипертелоризм, «карпий» рот, короткая уздечка языка, зубы Гетчинсона, короткая шея, симптом двузубца на стопах. Черепные нервы: глазные щели OD=OS, взор кратковременно фиксирует и прослеживает, зрачки OD=OS, фотореакция ослаблена, сглаженность правой носогубной складки, слух снижен, мелкоразмашистый нистагм, периодически с ротаторным компонентом, псевдобульбарный синдром.
Рефлекторно-двигательная сфера: двигательная активность резко снижена. Голову не держит, поворачивается на бок, не сидит, не ходит. Мышечная сила в конечностях снижена до 2-3 баллов. Мышечный тонус изменен: на фоне выраженного гипертонуса по спастическому типу периодически отмечаются изменения по типу «зубчатого колеса», больше в руках. Сухожильные рефлексы высокие с расширением рефлексогенных зон, D>S. Патологические кистевые и стопные рефлексы. Дистонические атаки. Ярко выраженные «стартл-рефлексы»: неожиданный звук, прикосновение вызывают стереотипную двигательную реакцию - резкое вздрагивание.
Высшие когнитивные функции: грубое нарушение психического развития - мало интересуется окружающим, однако узнает мать, смеется, сохранен ритм сна и бодрствования, дает знать о мокрых пеленках. Речевое развитие: певучее гуление с цепочками звуков, короткие лепетные звуки.
Данные дополнительных методов исследования
Ликворограмма: спинномозговая жидкость бесцветна, прозрачна, цитоз 1 мм3, белок 0,71 г/л.
Компьютерная ЭЭГ: грубые огранические изменения биоэлектрической активности, ирритативные общемозговые изменения биоэлектрической активности. Очаг грубой d-волновой активности с проекцией в задних отделах левого полушария. По данным в динамике, нарастание представленности и амплитуды d-колебаний, больше в левом полушарии, а также в затылочных отделах обоих полушарий, повышение ирритации в левой височной области.
KT головного мозга: на серии сагиттальных томограмм (6 мес, 1 год 1 мес, 2,5 года, 3 года) определяется диффузное изменение рисунка белого вещества больших полушарий мозга, ствола и гемисфер мозжечка. В динамике отмечается умеренное расширение III и боковых желудочков, IV желудочка, выраженное расширение конвекситальных субарахноидальных щелей в лобно-височных областях с двух сторон (рис. 1-3 ,
, ,
, ). В возрасте 3,5 лет на серии контрольных и аксиальных томограмм сохраняются ранее выявленные изменения, в режиме сканирования FLAIR у задних рогов боковых желудочков выявляются множественные мелкие кисты (рис. 4
). В возрасте 3,5 лет на серии контрольных и аксиальных томограмм сохраняются ранее выявленные изменения, в режиме сканирования FLAIR у задних рогов боковых желудочков выявляются множественные мелкие кисты (рис. 4  ).
).
Офтальмологическое исследование: OU спокойны; нистагм; подвижность глазных яблок полная. Глазное дно: диски зрительных нервов бледные, контуры четкие, артерии узкие, вены нормального калибра. Макулярная зона и периферия без особенностей. Диагноз: частичная атрофия зрительных нервов.
Зрительные вызванные потенциалы: органические изменения OU. Патология проводящих путей OU. Острота зрения по зрительным вызванным потенциалам равна 0,02-0,04.
Стволовая аудиограмма: пороги слуховой чувствительности слева в пределах нормы, справа порог снижен и соответствует тугоухости II степени по типу нейросенсорной.
Большие трудности в работе детского невролога представляет клиническая диагностика нейродегенеративных заболеваний ввиду их редкости, а также неспецифичности и «диффузности» симптомов в ранних стадиях. Такие симптомы заболевания, как вялое сосание, нарушение фиксации взора, тремор, повышенная возбудимость, нарушение мышечного тонуса, судороги, апноэ, характерны для разных заболеваний генетической и негенетической природы [29]. Вероятнее всего, поэтому в нашем случае ребенок наблюдался в разных стационарах со следующими диагнозами: перинатальное поражение ЦНС, гипертензионный синдром, гидроцефалия, детский церебральный паралич.
Состояния, составляющие группу лейкодистрофий, относятся к дегенеративным заболеваниям нервной системы, но сопровождаются значительным нарушением процесса миелинизации. Основными критериями, позволяющими заподозрить лейкодистрофию, являются: дебют в определенном возрасте, наличие периода нормального развития, преимущественное поражение двигательных отделов нервной системы (пирамидные, экстрапирамидные, мозжечковые расстройства), нарушение поведения, деменция, а также признаки демиелинизации при КТ и МРТ головного мозга [24]. Неврологическая симптоматика имеет симметричный и прогредиентный характер. На глазном дне обычно обнаруживается атрофия зрительных нервов. Характерна белково-клеточная диссоциация в спинномозговой жидкости. МРТ высокочувствительна к дисмиелогенным нарушениям, но не очень специфична в плане дифференциальной диагностики. При всех этих патологических состояниях на Т2-взвешенных томограммах и томограммах, отражающих протонную плотность, видны гиперинтенсивные поля в белом веществе. МРТ-признаки плохо коррелируют с тяжестью клинического течения заболевания. Роль этого исследования заключается не в установлении высокоспецифичного диагноза или оценке тяжести процесса, а в дифференциальной диагностике с заболеваниями, имеющими сходную симптоматику [5].
В то же время имеются определенные клинические особенности и биохимические показатели, которые позволяют заподозрить определенную форму лейкодистрофий: наследственный анамнез, фенотип, дебют заболевания, особенности его клиники и течения, вовлечение других органов и систем [8]. Основную роль в дифференциальной диагностике лейкодистрофий играют биохимические и молекулярно-генетические методы.
М. Adachi и соавт. описали три клинические формы БК в зависимости от возраста дебюта: врожденную, инфантильную и ювенильную [6, 7, 12, 29].
Наиболее хорошо изучена клинически инфантильная форма заболевания. На первом месяце у детей часто отмечаются нарушение фиксации взора, повышенная возбудимость, вялое сосание, слабый зрительный контакт «глаза в глаза», судороги, диффузная мышечная гипотония [29]. Основные клинические признаки БК становятся очевидными к 3 мес жизни. Важнейшими из них являются выраженная мышечная гипотония, неспособность удерживать голову в вертикальном положении, в дальнейшем трансформация диффузной мышечной гипотонии в спастичность. Макроцефалия является характерным симптомом заболевания, но в раннем возрасте окружность головы может оставаться в пределах нормы. Патологический прирост окружности головы сохраняется после 6 мес жизни и может сочетаться с задержкой закрытия большого родничка [25]. B возрасте 1 года окружность головы может превышать 90 центилей. По данным G. Gascon и соавт. [13], мышечная гипотония, сопровождающаяся снижением двигательной активности, в последующем сменяется спастическим гипертонусом, что указывает на вовлечение в патологический процесс пирамидной системы. Наблюдается повышение сухожильных рефлексов, появляются патологические кистевые и стопные рефлексы, и по мере прогрессирования болезни развивается децеребрационная или декортикационная ригидность. Часто наблюдаются высокие «стартл-рефлексы». По мере развития заболевания нарастает нарушение психомоторного развития и, кроме того, у 50% больных присоединяются генерализованные тонико-клонические судороги. Судороги и атрофия зрительных нервов обычно развиваются на втором году жизни, к этому же возрасту увеличение окружности головы достигает плато [27]. Пациенты с БК могут быть легко возбудимы или, наоборот, повышенно сонливы.
Прогрессирование заболевания приводит к нарушениям работы желудочно-кишечного тракта, обусловливающим трудности вскармливания и гипотрофию. Большинство пациентов умирают в первой декаде жизни. При более поздних сроках манифестации заболевания продолжительность жизни увеличивается [13, 25, 27].
На основании сочетания в клинической картине заболевания макроцефалии, задержки психомоторного развития, генерализованных тонических судорог, высоких «стартл-рефлексов», спастического тетрапареза, прогрессирующего характера течения процесса, а также выявления очагов демиелинизации при КТ и МРТ у нашего пациента было заподозрено дегенеративное заболевание нервной системы. Дифференциальная диагностика проводилась со следующими заболеваниями: болезнью Краббе (глобоидно-клеточная лейкодистрофия), GM2 ганглиозидозы (тип 1 и 2), болезнью Александера, мегалэнцефалией и лейкодистрофией (болезнь Ван дер Кнаапа, или вакуолизирующая лейкоэнцефалопатия), синдромом Сотоса.
Сочетание у нашего пациента описанных выше симптомов позволило предположить БК или болезнь Александера. На уровне клинического осмотра различить эти заболевания достаточно сложно. Сроки дебюта заболеваний и его основные симптомы сходны, однако применение методов нейровизуализации позволило с наибольшей вероятностью установить правильный диагноз.
При проведении МРТ головного мозга в динамике нами была обнаружена прогредиентность выявленных при первичном исследовании изменений в белом веществе головного мозга. Гиподенсные по данным КТ и гиперинтенсивные на T2-взвeшенныx МР-изображениях зоны увеличились: если при начальном исследовании они занимали незначительные регионы перивентрикулярных отделов, то при последнем достигали степени субтотального поражения белого вещества конечного мозга.
Для точной верификации диагноза мы определили концентрацию N-ацетиласпартата в моче методом ВЭЖХ. Было выявлено значительное повышение его уровня: 2700 ммоль/моль креатинина (норма - до 700 ммоль/моль креатинина). На основании результатов этого биохимического исследования был установлен диагноз БК.
При проведении ДНК-диагностики в 6-м экзоне гена (ASPA) аспартоацилазы была обнаружена мутация А305Е в гетерозиготном состоянии. В настоящее время проводится поиск второй мутации для разработки метода прямой ДНК-диагностики в данной семье.
Таким образом, на основании ведущего клинического синдрома, длительного наблюдения за ребенком, результатов КТ и МРТ в динамике, данных биохимических исследований (повышение экскреции N-ацетиласпартата в моче), обнаружения мутации гена нами был установлен диагноз болезни Канавана.
Впервые в России этот диагноз подтвержден на биохимическом и молекулярно-генетическом уровне.