Лимфангиолейомиоматоз: современный взгляд на проблему
Н.И. ШВЕЦ, д. мед. н., профессор; Т.М. БЕНЦА, к. мед. н., доцент; В.В. СТАНИШЕВСКИЙ
/Национальная медицинская академия последипломного образования им. П.Л. Шупика, Киев/
Лимфангиолейомиоматоз (лейомиоматоз) (ЛАМ) - это редкая патология, возникающая у женщин детородного возраста 18-50 лет; характеризуется прогрессирующей одышкой, пневмо-, хилотораксом и кровохарканьем. В основе ЛАМ - диссеминиро-ванный патологический процесс, характеризующийся опухолевидным разрастанием гладкомышечных волокон по ходу мелких бронхов, бронхиол, стенок кровеносных и лимфатических сосудов легких с последующей мелкокистозной трансформацией легочной ткани. Основное проявление ЛАМ - прогрессирующая дыхательная недостаточность.
Диффузный ЛАМ (диффузный лейомиоматоз легких, легочной лейомиоматоз, фибролейомиоматозная гамартома) принадлежит к числу редко встречающихся заболеваний. Первое описание ЛАМ датировано 1937 годом. С этого времени зарегистрировано немногим более 100 случаев ЛАМ. Однако за последние 5 лет отмечается резкий рост данной патологии в странах Европы.
Этиология и патогенез
Этиология ЛАМ остается неизвестной. Предполагают гормо-нозависимость (эстрогенозависимость) заболевания. Косвенно эта версия подтверждается тем, что ЛАМ встречается преимущественно у женщин репродуктивного возраста, крайне редко -у мужчин. Обостряется заболевание во время беременности, в предменструальном периоде, а стабилизация процесса отмечается в постменопаузе. Сочетание ЛАМ легких с лейомиомой матки также указывает на важную роль эндокринных нарушений в развитии болезни. Не исключено, что возникновение заболевания связано с иммунными нарушениями. Имеются также данные о том, что определенную роль в развитии ЛАМ играют генетические нарушения в белках, вовлеченных в синтез катехоламинов.
Существующие теории возникновения заболевания не объясняют в полной мере его причину. Наибольшее клиническое подтверждение находит теория гормональных нарушений. По другим данным, в основе заболевания лежит асинхронная мышечная пролиферация в легких, матке и, возможно, в мышцах другой локализации. Еще одна теория основана на том, что лейомиома-тозные узлы возникают через 1-20 лет после удаления матки по поводу фибромиомы, что связано с эмболией сосудистого русла гладкомышечными клетками.
Клиника
В начальной стадии клинические проявления могут отсутствовать. Длительное время заболевание протекает бессимптомно. ЛАМ часто обнаруживается случайно как диффузное или мелкоузловое поражение легочной ткани при рентгенологическом исследовании органов грудной клетки. Пациенты обращаются за помощью при появлении прогрессирующей одышки вследствие развивающейся обструкции дыхательных путей и снижения диффузионной способности легочной ткани.
Основные клинические проявления:
одышка, вначале беспокоит только при физической нагрузке, в дальнейшем становится постоянной;
боли в грудной клетке, усиливающиеся при дыхании;
кровохарканье (непостоянный симптом);
рецидивирующий спонтанный пневмоторакс - наблюдается у 1/2-1/3 больных, проявляется внезапной интенсивной болью в грудной клетке, одышкой, отсутствием везикулярного дыхания и тимпаническим оттенком перкуторного звука на стороне поражения;
хилоторакс - скопление хиллезной жидкости в плевральной полости (с одной или обеих сторон). При развитии хилоторакса усиливается одышка, появляется интенсивный тупой звук при перкуссии над областью выпота, дыхание в этом месте отсутствует; хиллезная жидкость накапливается вновь после ее удаления. Характерно, что развитие пневмо- и хилоторакса совпадает с менструацией;
хилоперикардит и хиллезный асцит развиваются по мере прогрессирования заболевания, их появление совпадает с менструальным циклом;
развитие легочного сердца - важнейшим симптомом является одышка, усиливающаяся при физической нагрузке; при выраженной легочной гипертензии она наблюдается и в покое. Характерными особенностями одышки являются отсутствие ортопноэ и уменьшение ее при использовании ингаляций кислорода. Больных беспокоят также выраженная слабость, сердцебиение, боли в области сердца. Кардиалгии обусловлены гипоксией, рефлекторным сужением коронарных артерий (пульмокоронарным рефлексом), уменьшением наполнения коронарных артерий при увеличении конечного диастолического давления в полости правого желудочка. Боли в области сердца носят постоянный характер и уменьшаются после ингаляций кислорода. Чрезвычайно характерен теплый диффузный серый цианоз, обусловленный артериальной гипоксемией. На фоне хронической гипоксии и гиперкапнии появляются постоянные головные боли, сонливость днем, бессонница ночью, потливость, снижается аппетит. При декомпенсации хронического легочного сердца развиваются ортопноэ, холодный акроцианоз, набухание вен, которое не уменьшается на вдохе, увеличение печени, симптом Плеша (надавливание на увеличенную болезненную печень вызывает набухание шейных вен), при тяжелой сердечной недостаточности возможно развитие отеков, асцита, гидроторакса.
Очаговая форма ЛАМ протекает бессимптомно и выявляется рентгенологически. В некоторых случаях заболевание принимает системный характер - лейомиомы развиваются в брюшной полости, забрюшинном пространстве, матке, кишечнике, почках. Ангиомиолипомы почек редко нарушают функцию почек, хотя иногда могут достигать больших размеров (более 10 см).
Активации заболевания способствуют беременность, роды, прием контрацептивов. Прогноз у таких больных, как правило, неблагоприятный. Летальный исход наступает в сроки от двух до 10-ти лет. Средняя продолжительность жизни больных составляет около 5 лет. Описаны случаи с летальным исходом через 17 лет. Непосредственная причина смерти - прогрессирующая дыхательная недостаточность.
Лабораторные методы диагностики
Общий анализ крови: существенных изменений нет. У некоторых больных отмечается эозинофилия, нередко увеличивается СОЭ, особенно при развитии пневмо-хилоторакса.
Общий анализ мочи: может наблюдаться незначительная протеинурия (симптом неспецифический и непостоянный).
Биохимическое исследование крови: иногда наблюдается гиперхолестеринемия, возможно увеличение уровня а2- и у-глобулинов, аминотрансфераз, общей лактатдегидрогеназы, ангиотензинпревращающего фермента
Исследование плевральной жидкости: хилоторакс чрезвычайно характерен для ЛАМ. Плевральная жидкость имеет следующие характерные особенности:
цвет молочно-белый;
мутность жидкости сохраняется после центрифугирования;
содержание триглицеридов выше 110 мг %;
содержит хиломикроны, которые выявляются при электрофорезе липопротеинов в полиакриламидном геле.
Инструментальные методы исследования
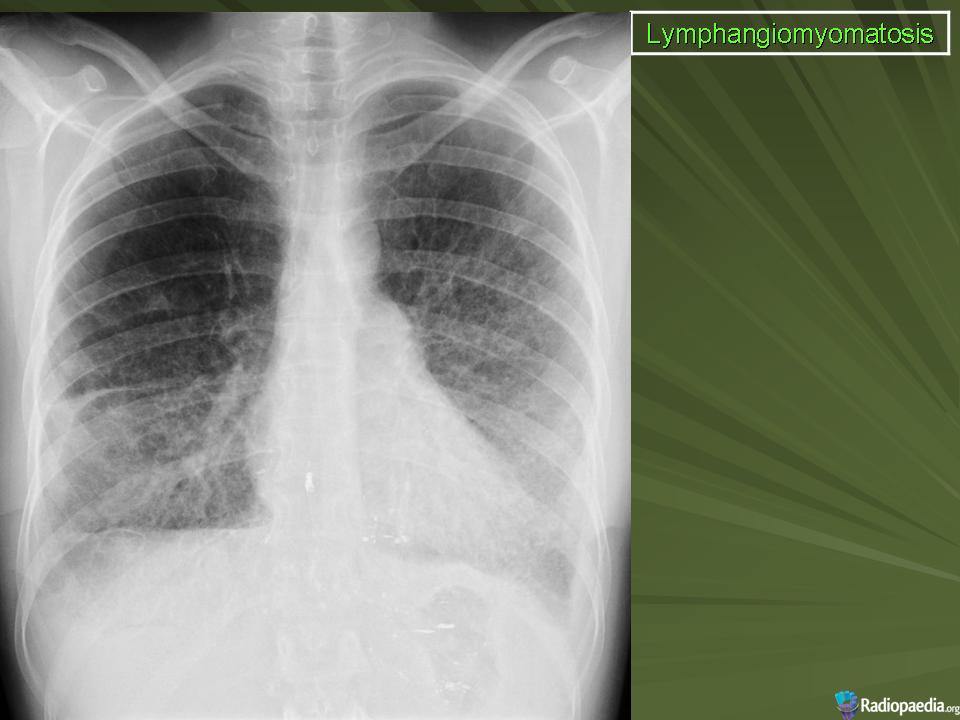
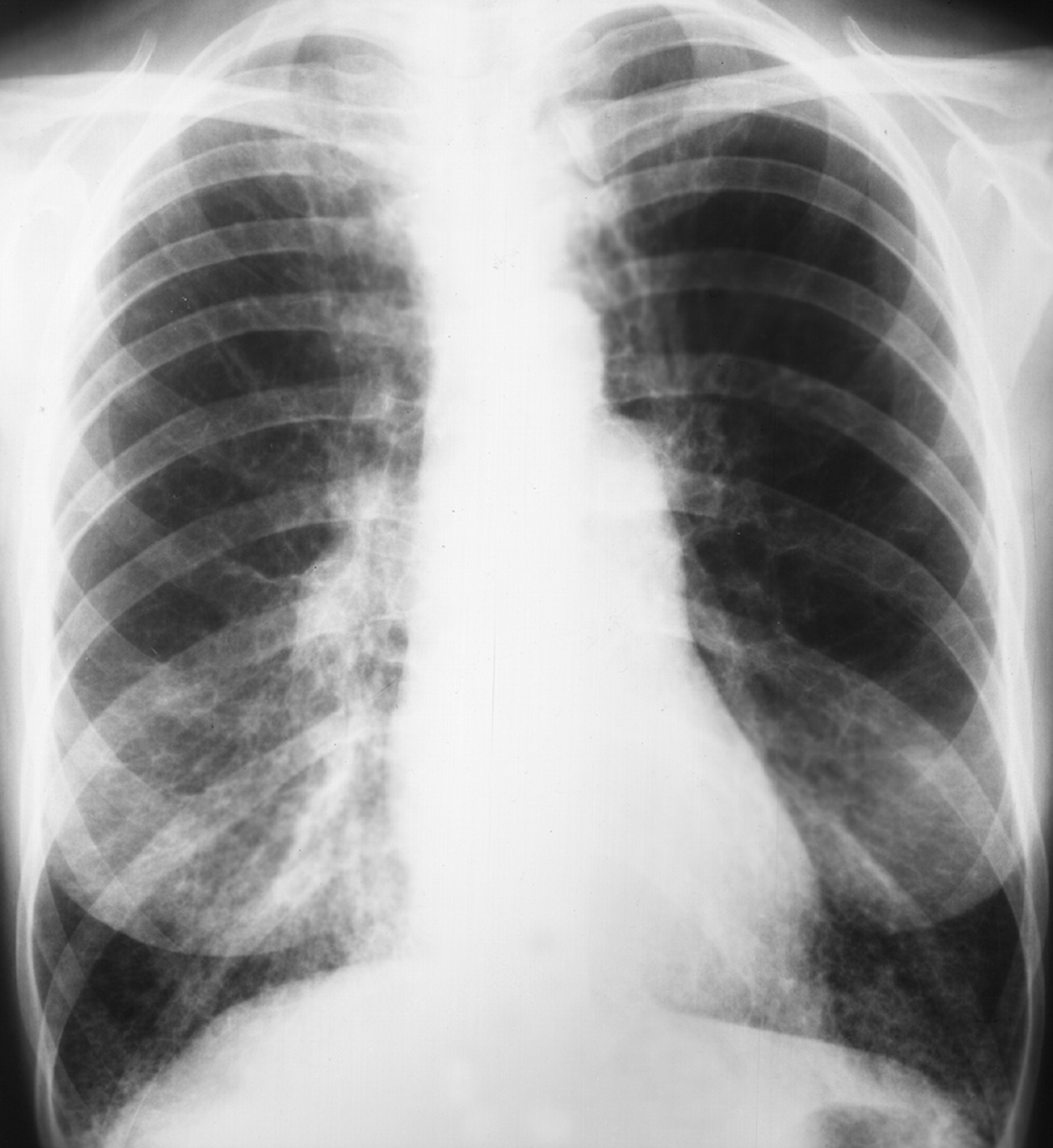
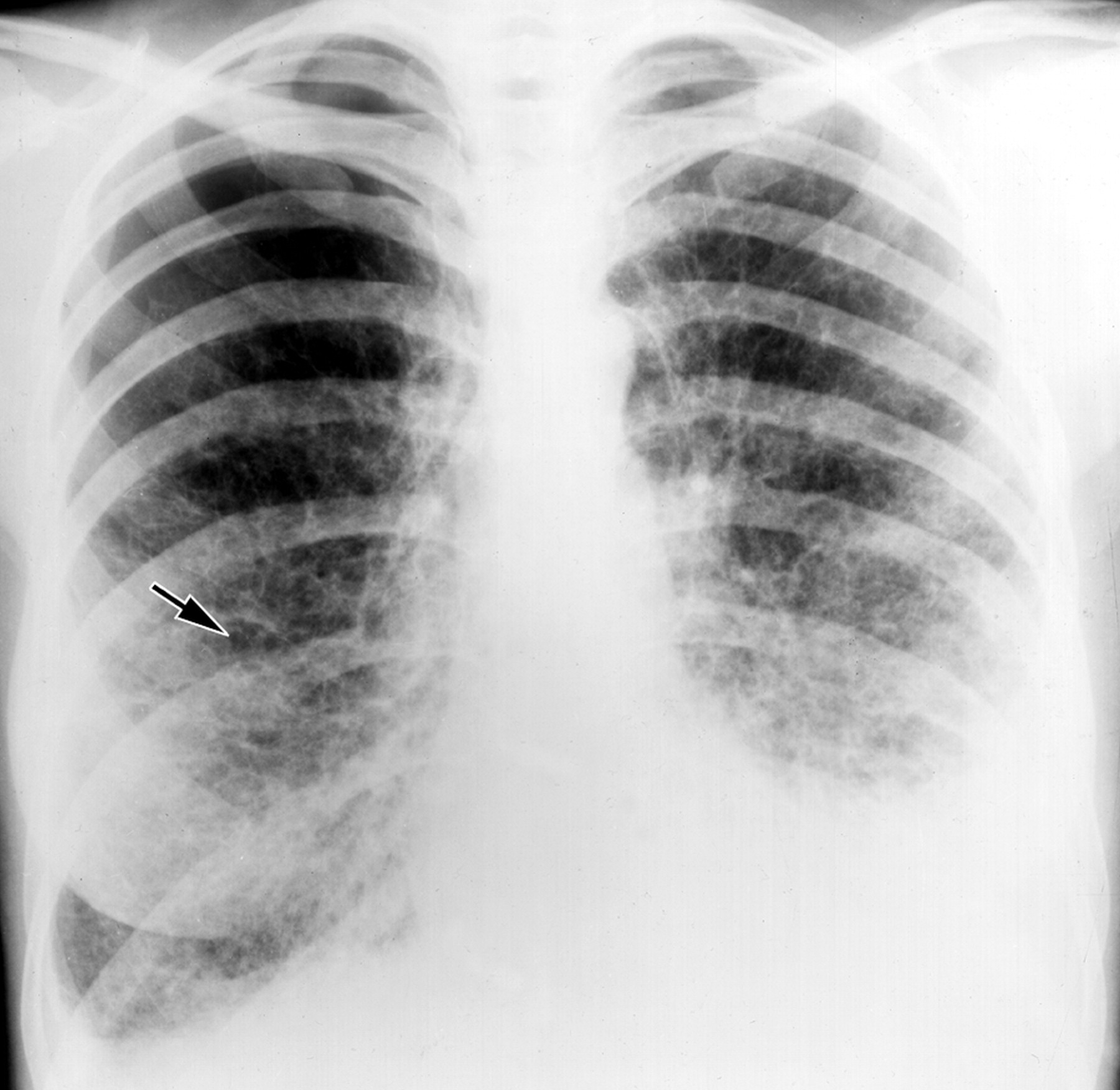
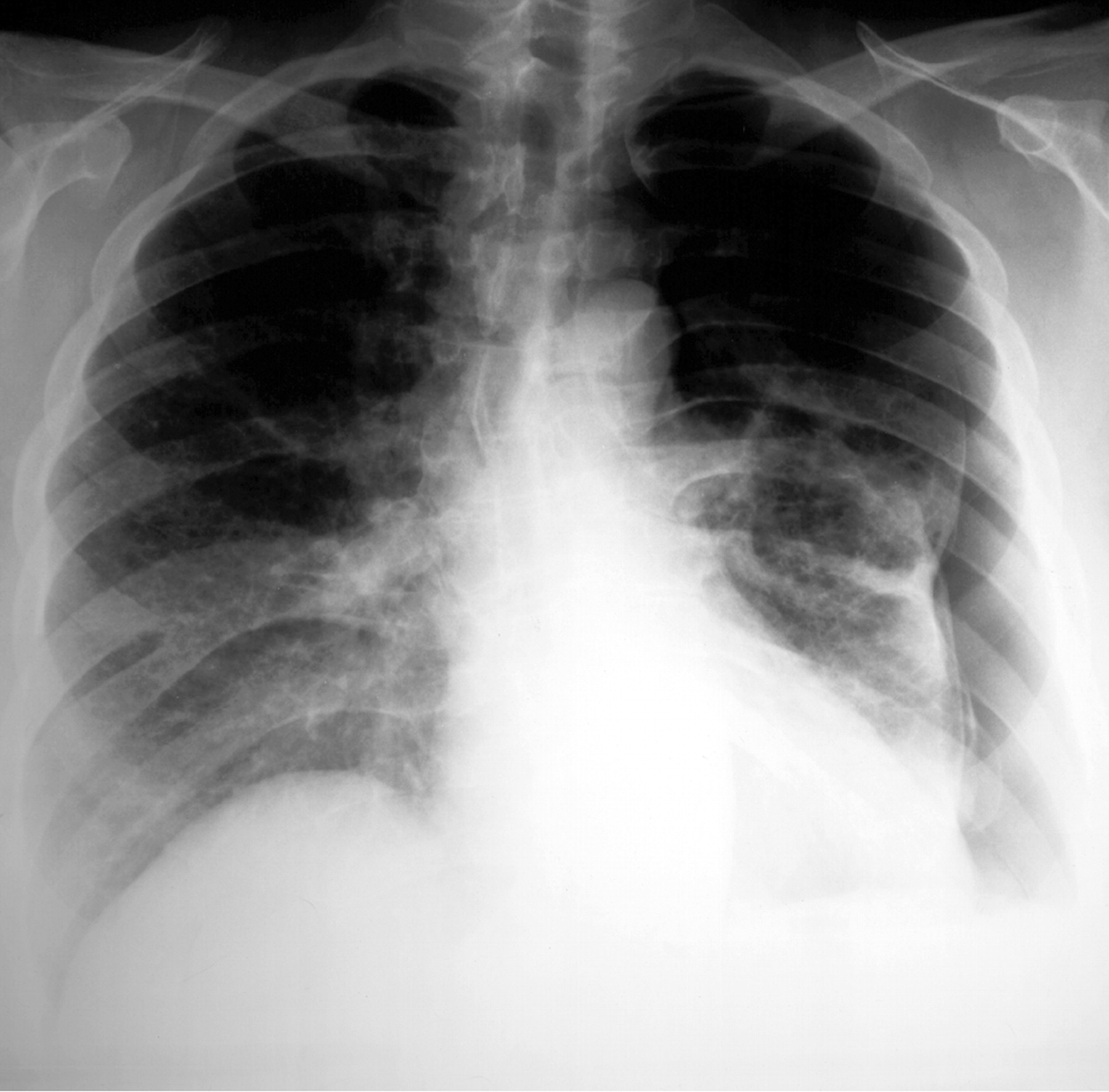
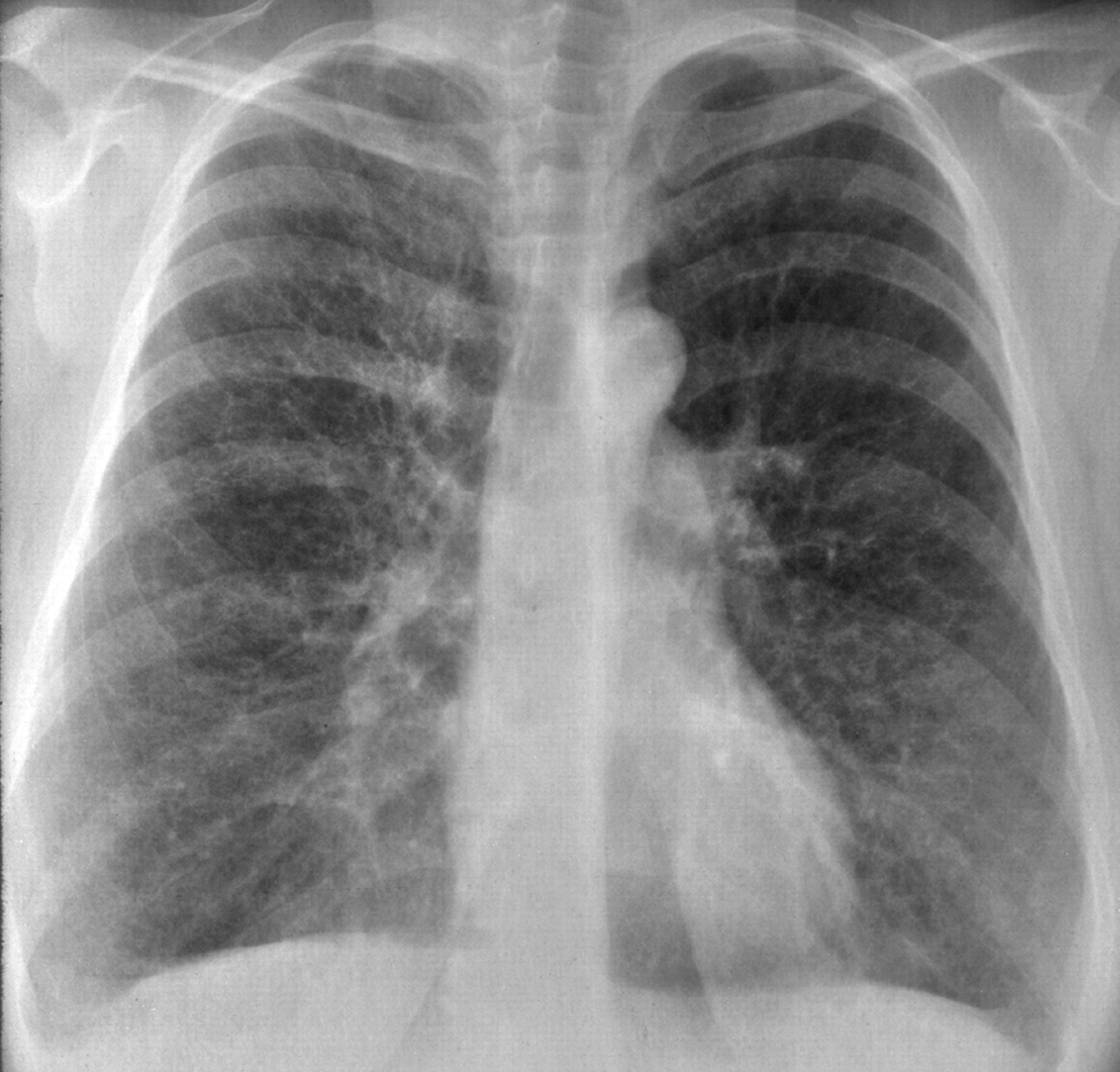
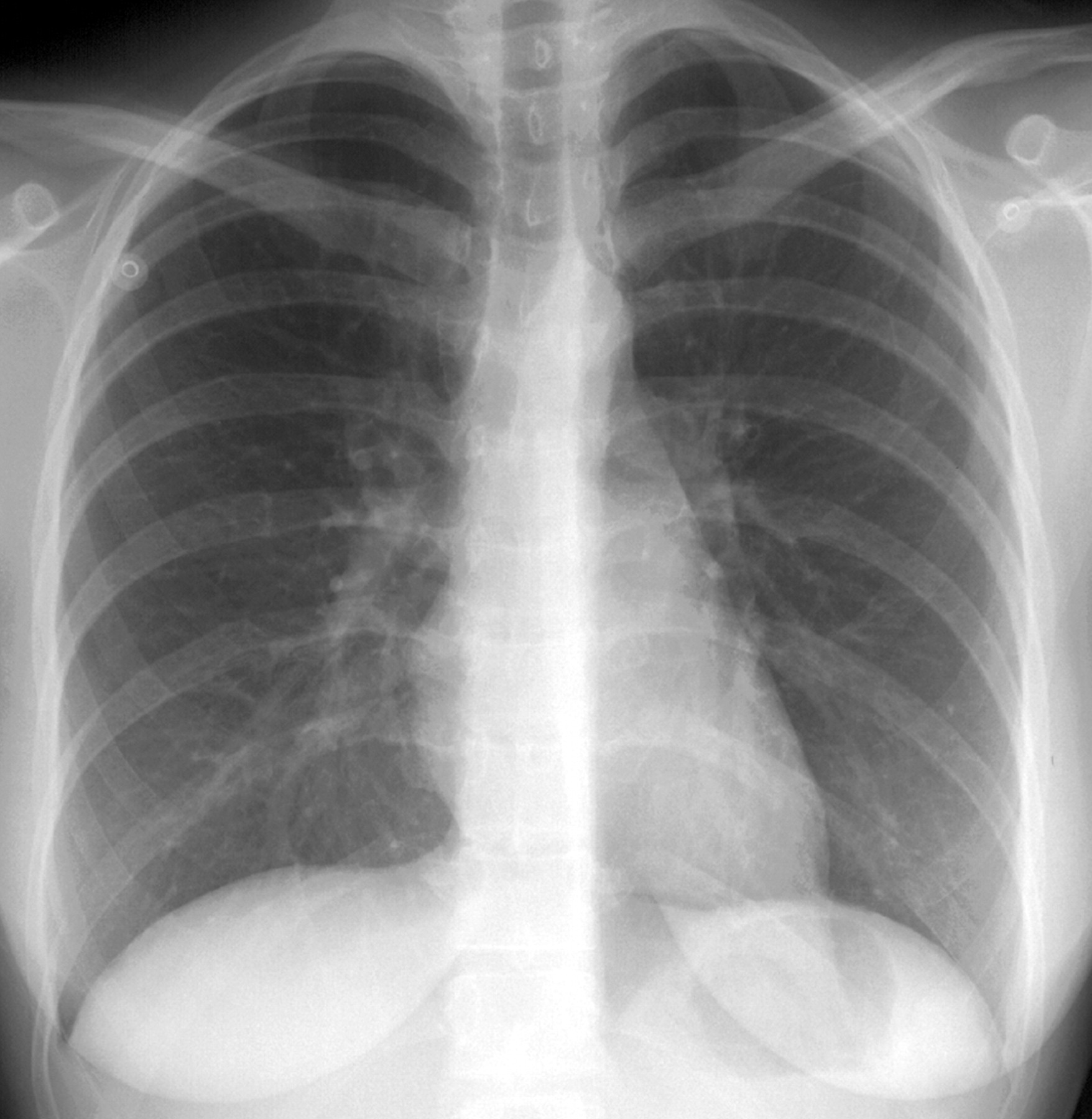
Рентгенологическое исследование легких. Главными рентгенологическими признаками ЛАМ легких на обычных рентгенограммах грудной клетки являются:
усиление легочного рисунка сетчатого характера;
увеличение объема легких.
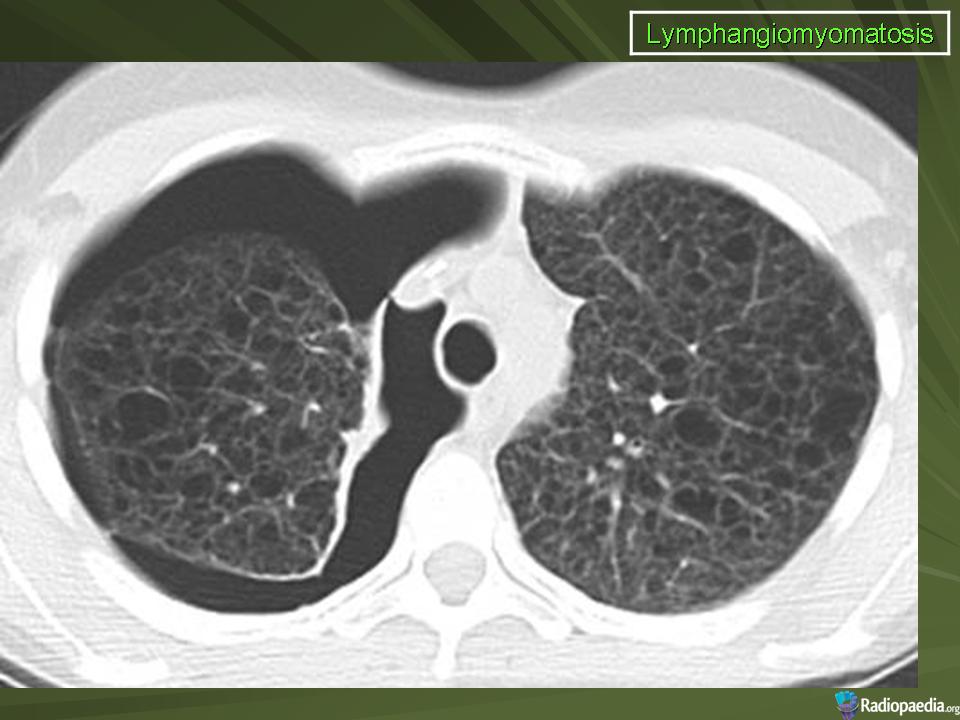
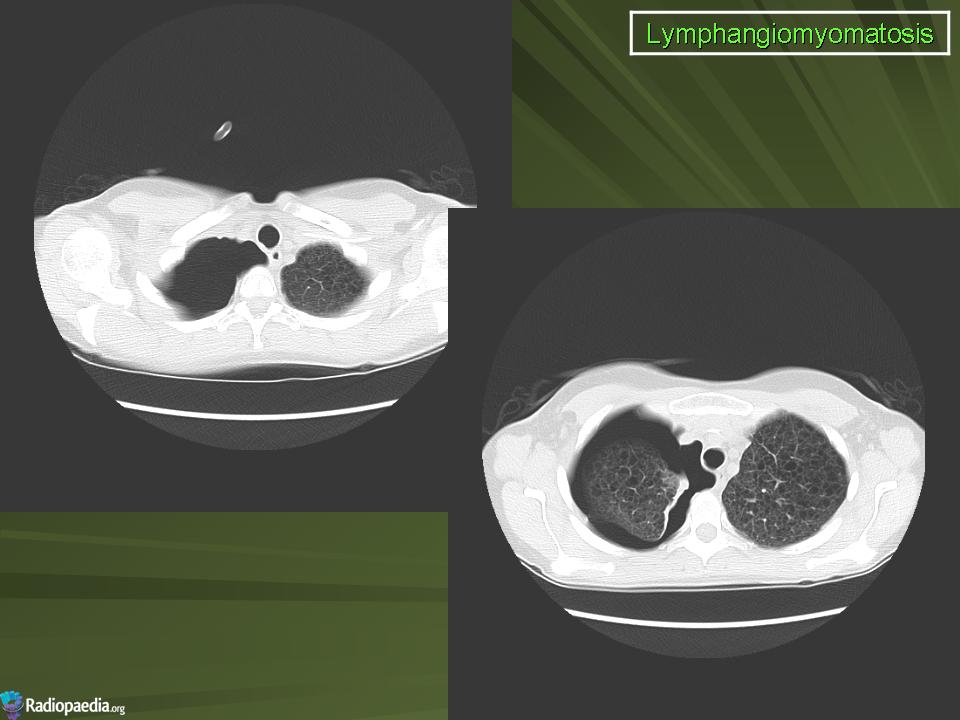
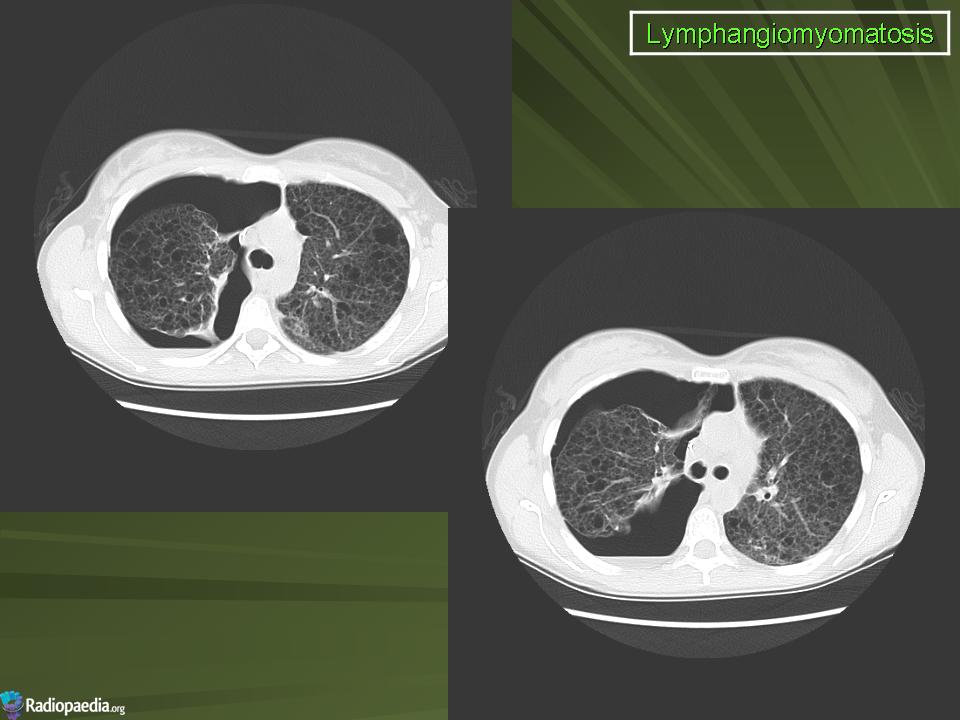
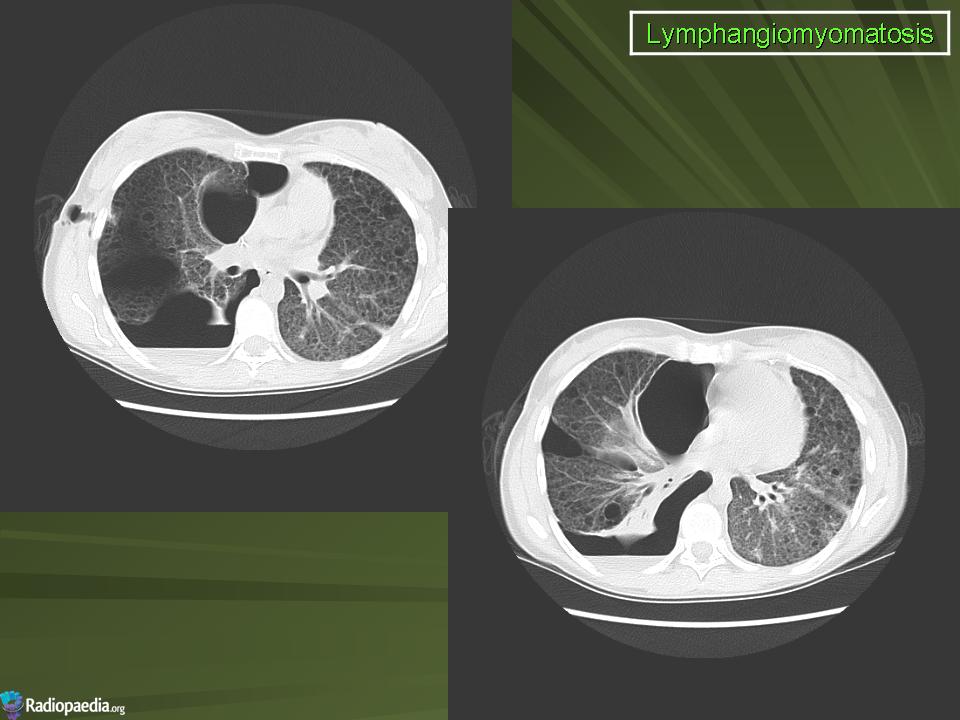
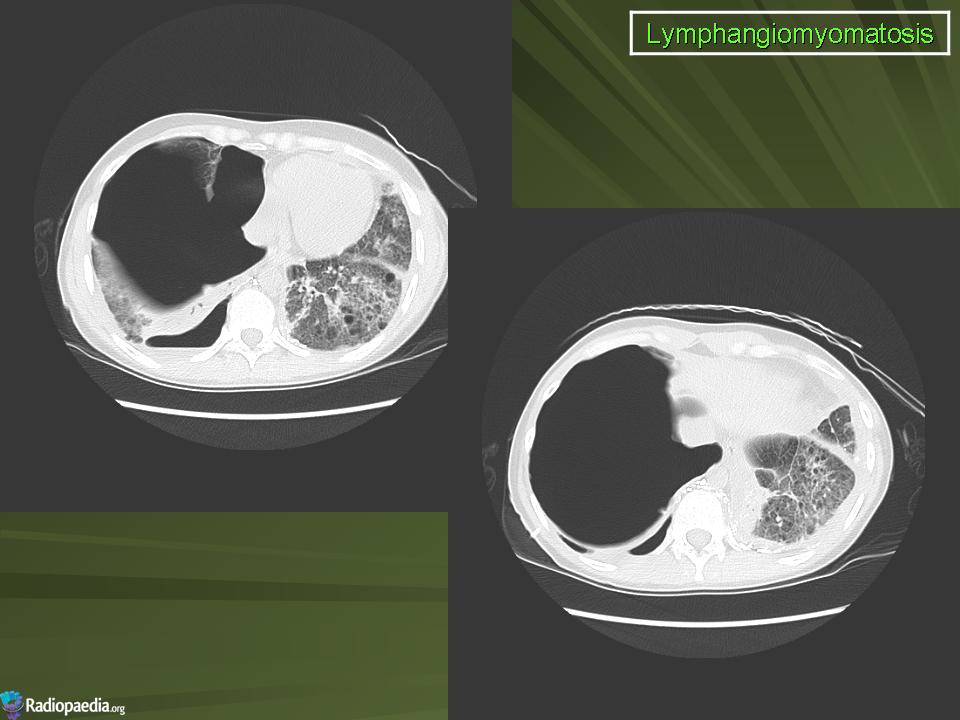
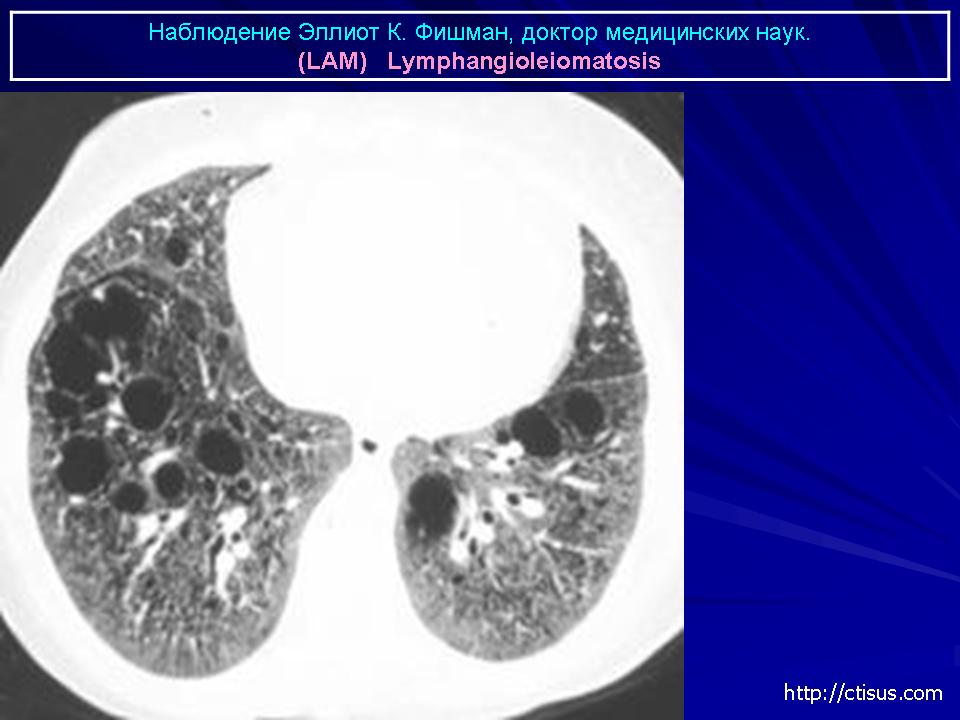
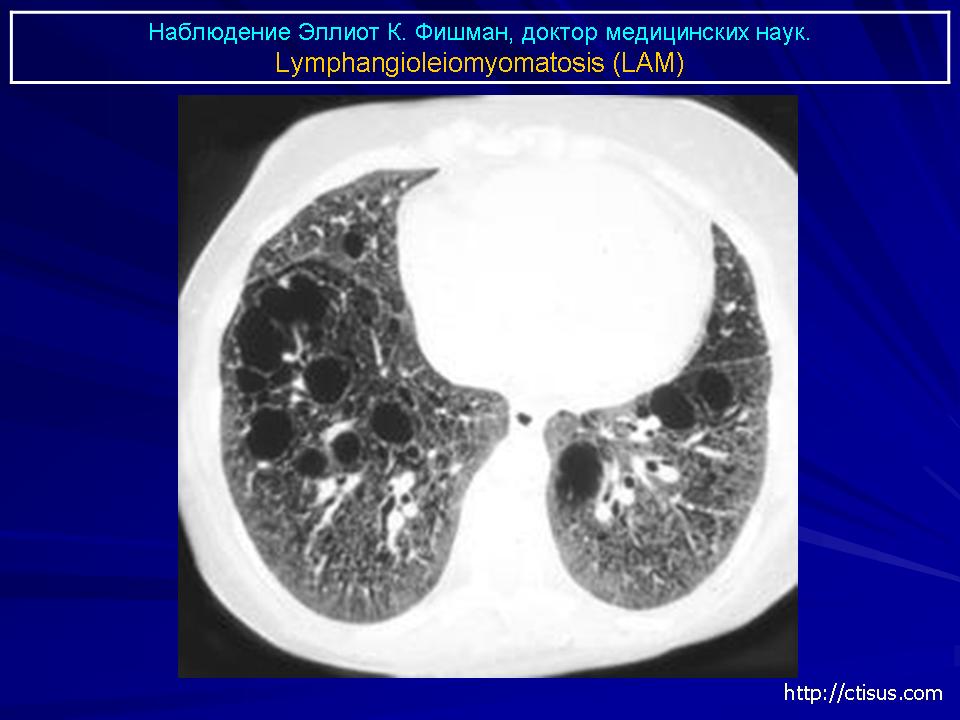
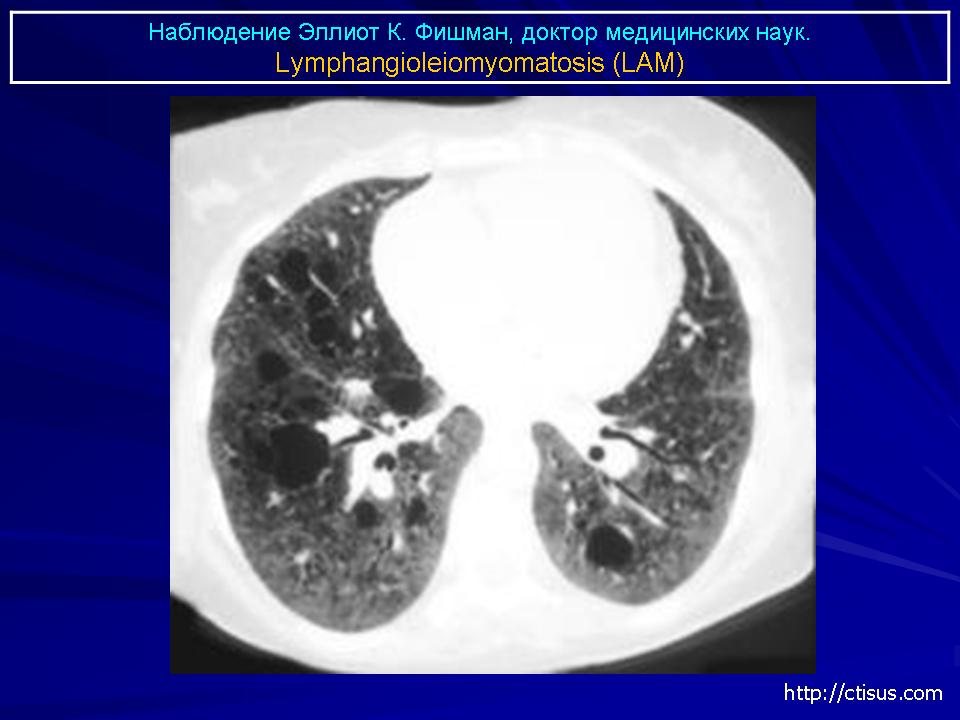
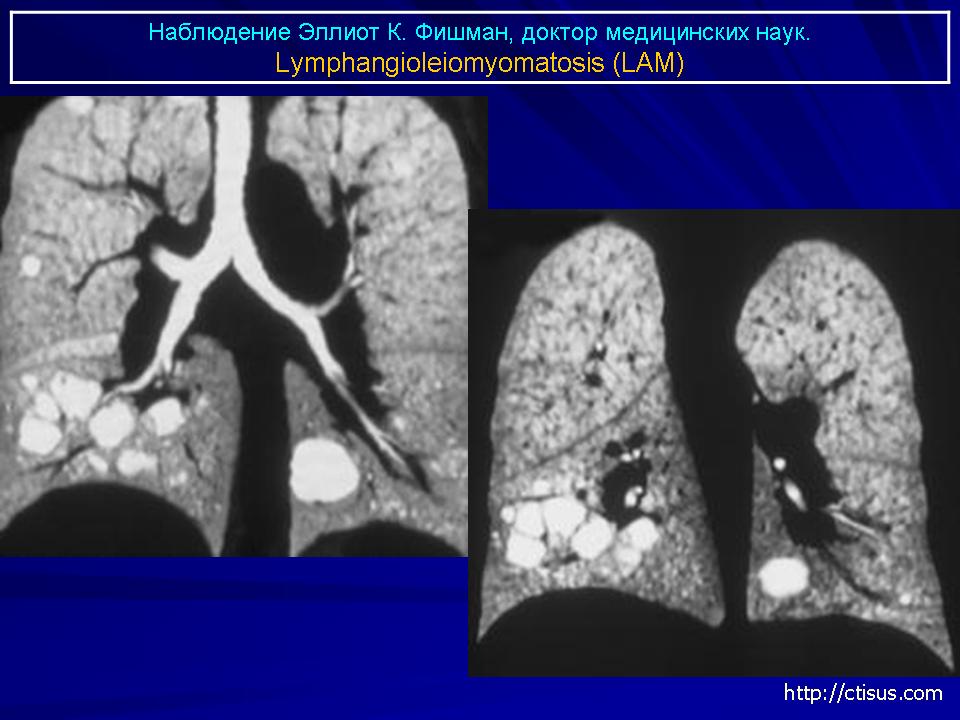
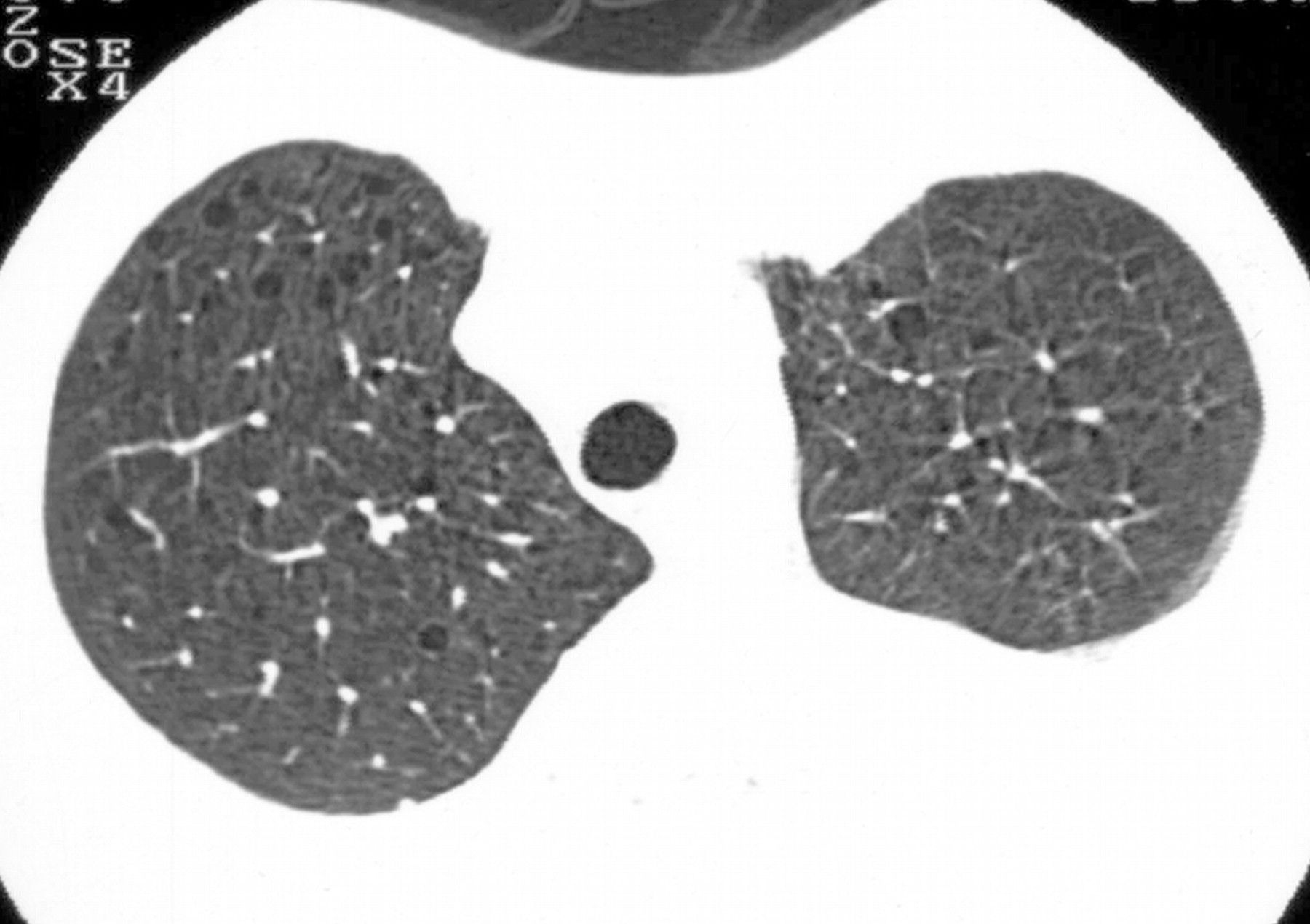
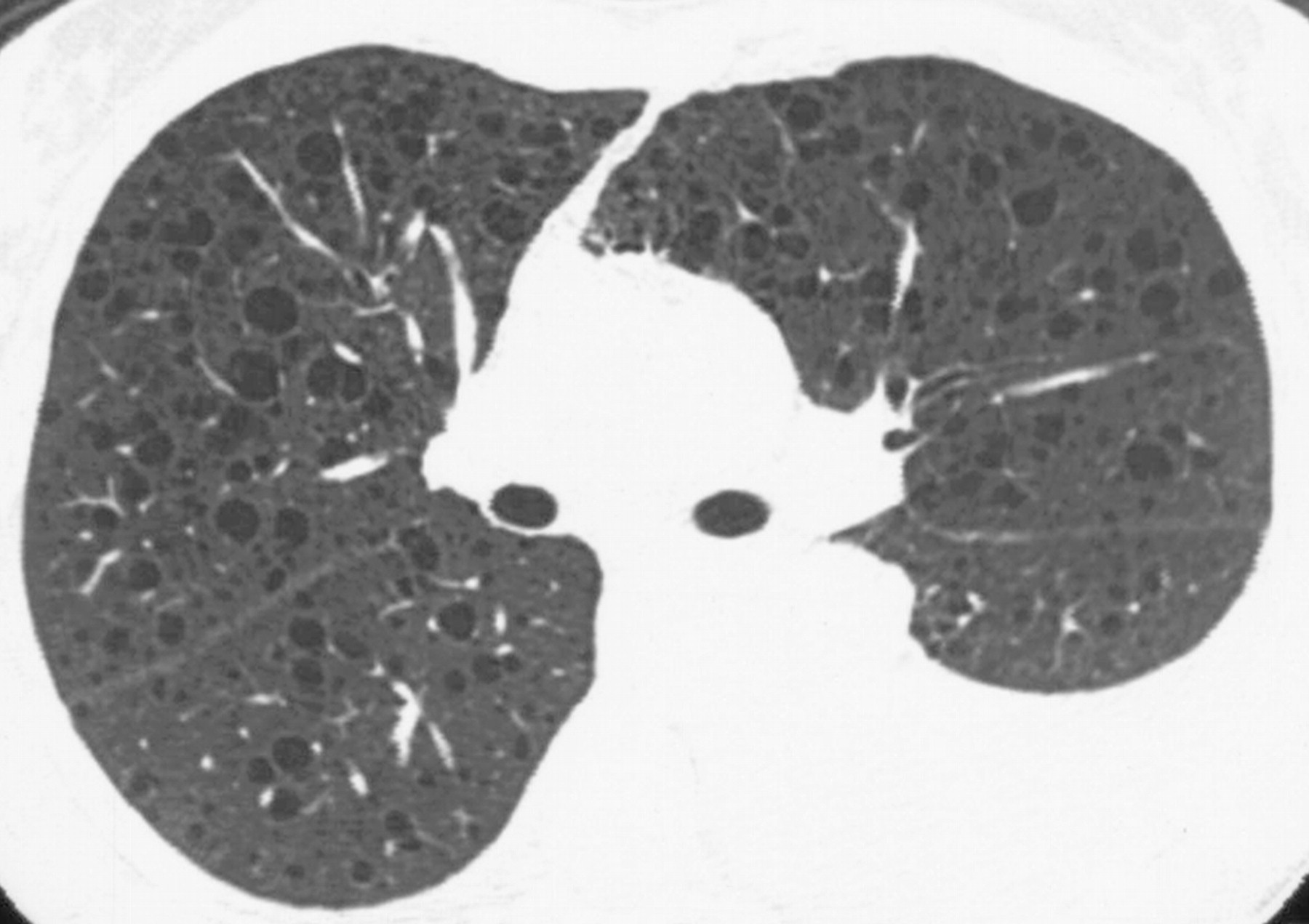
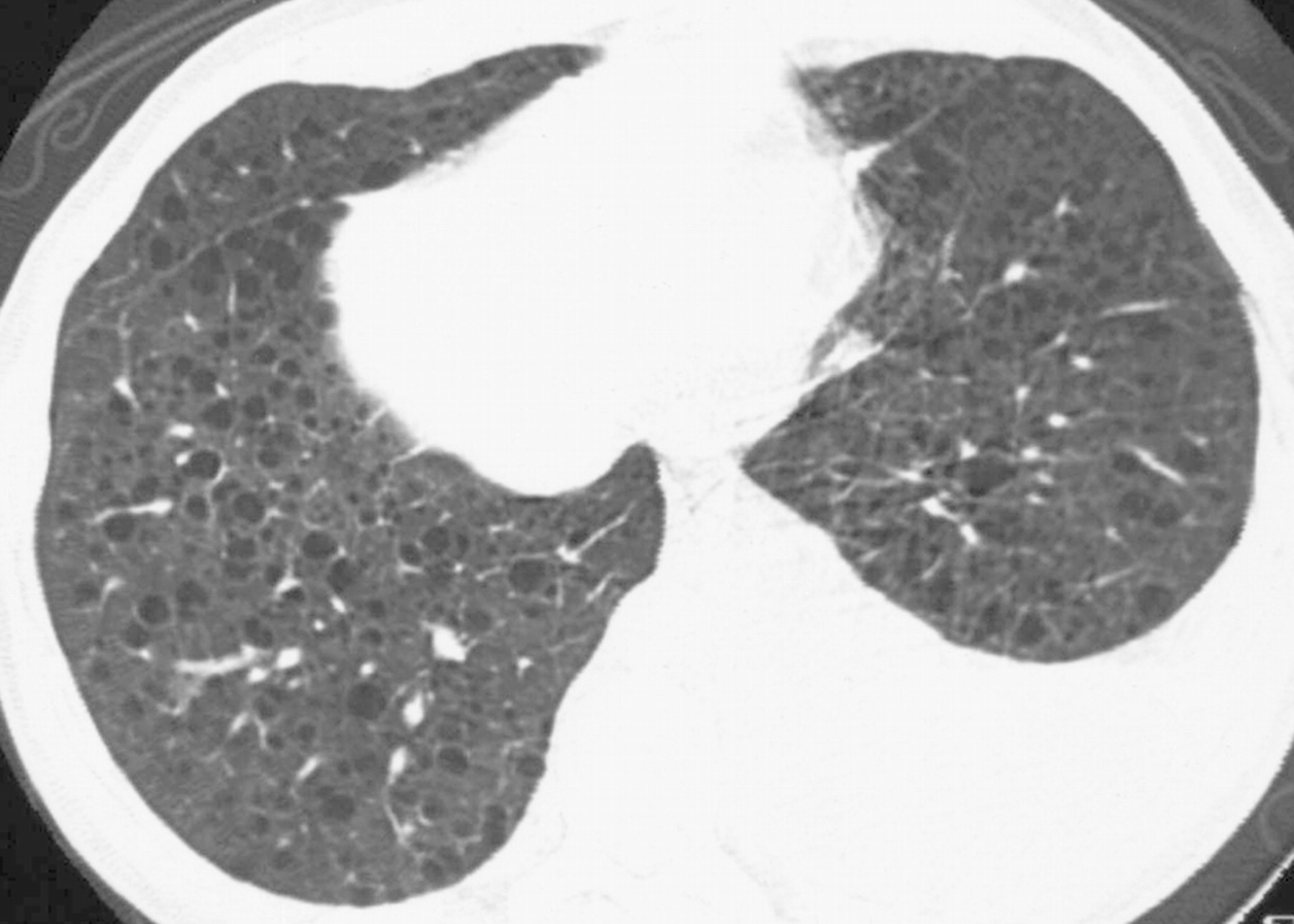
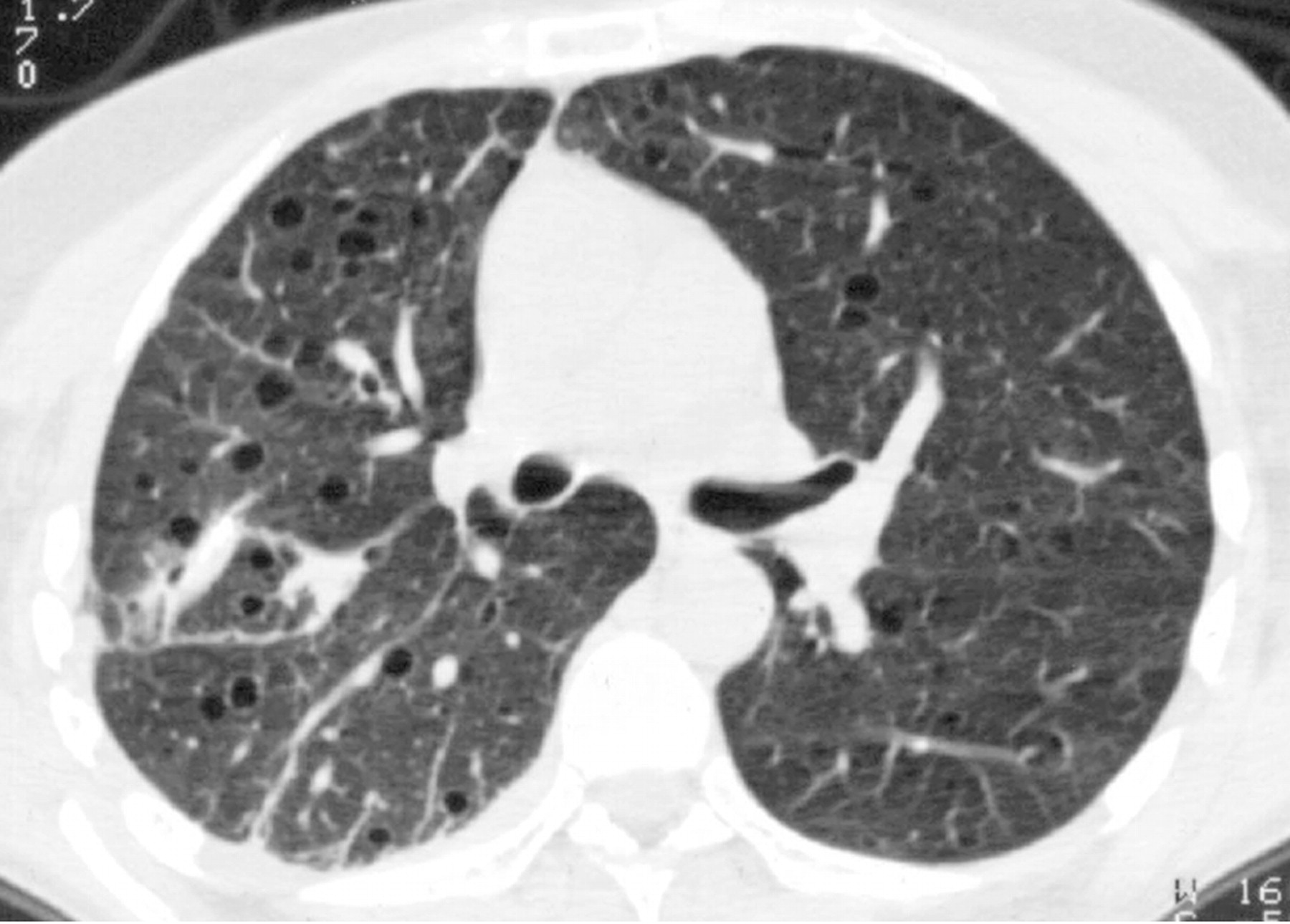
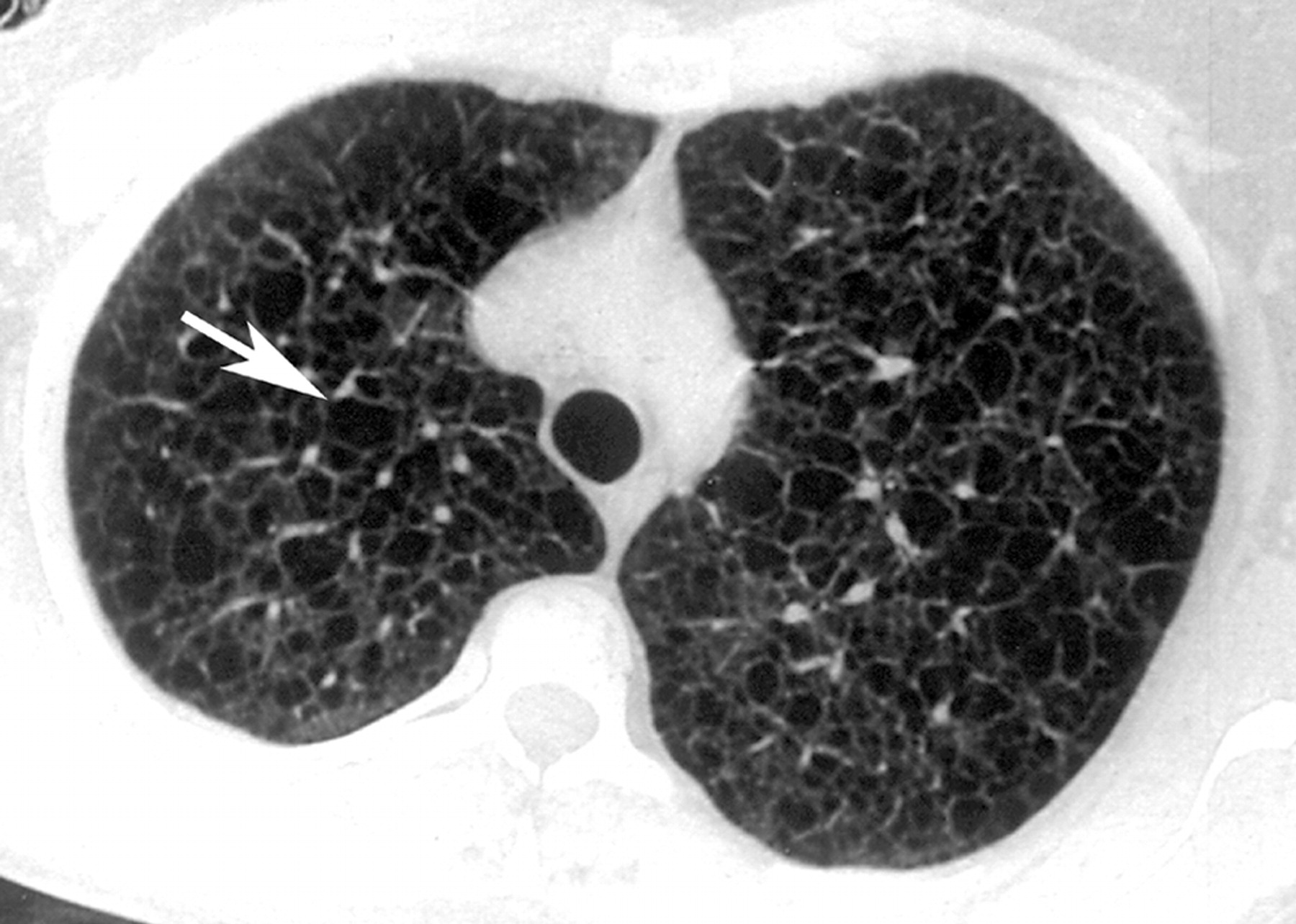
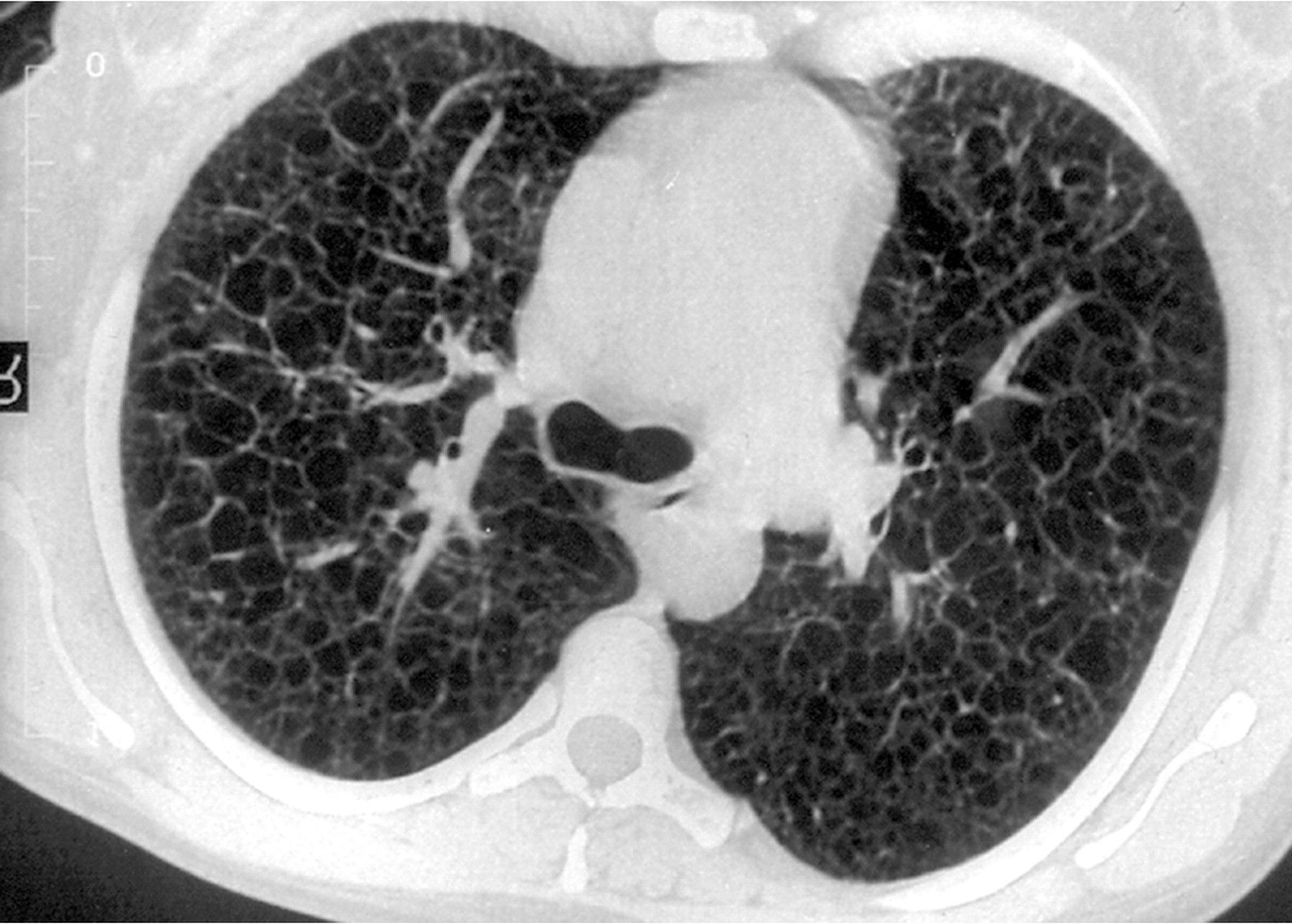
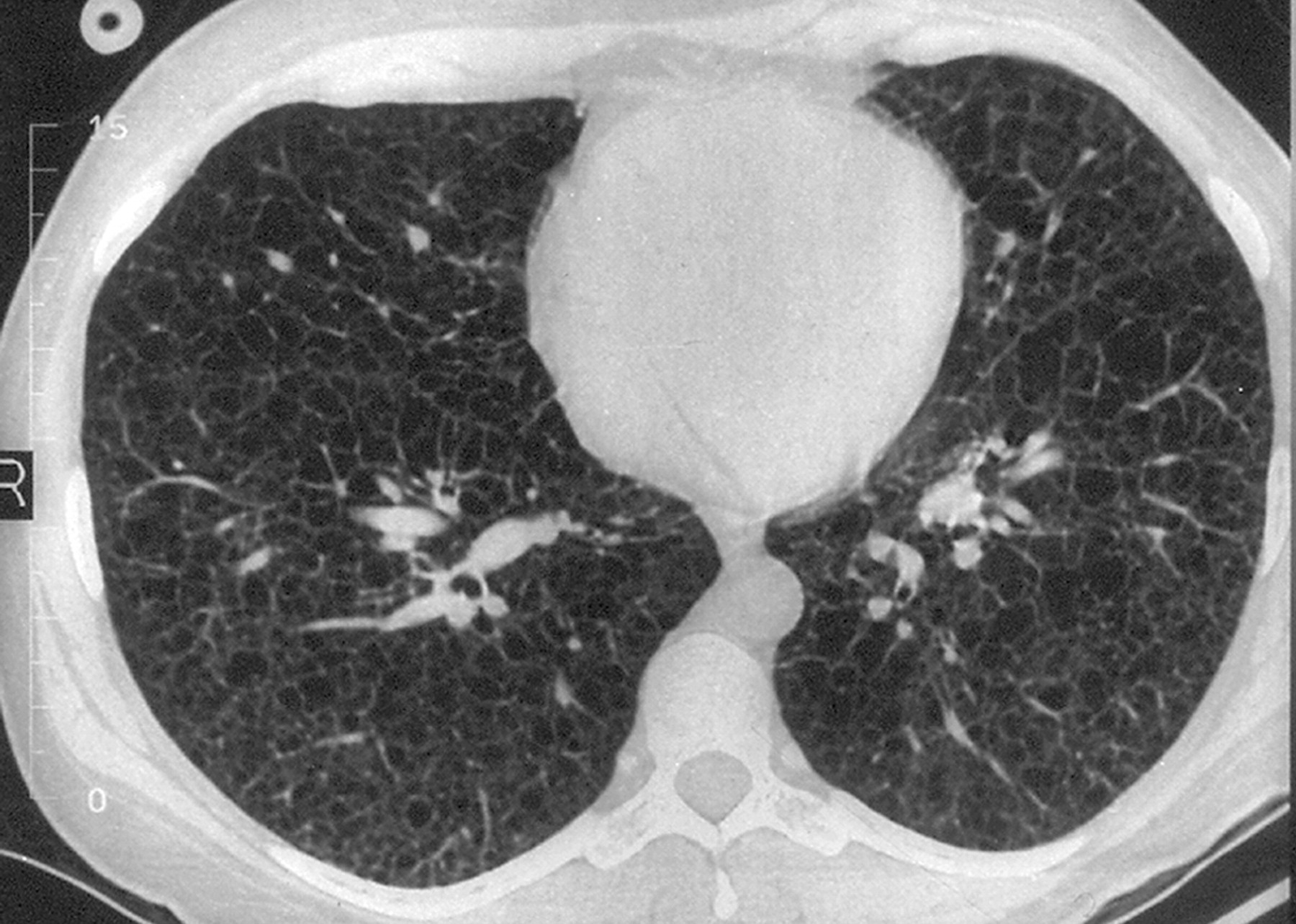
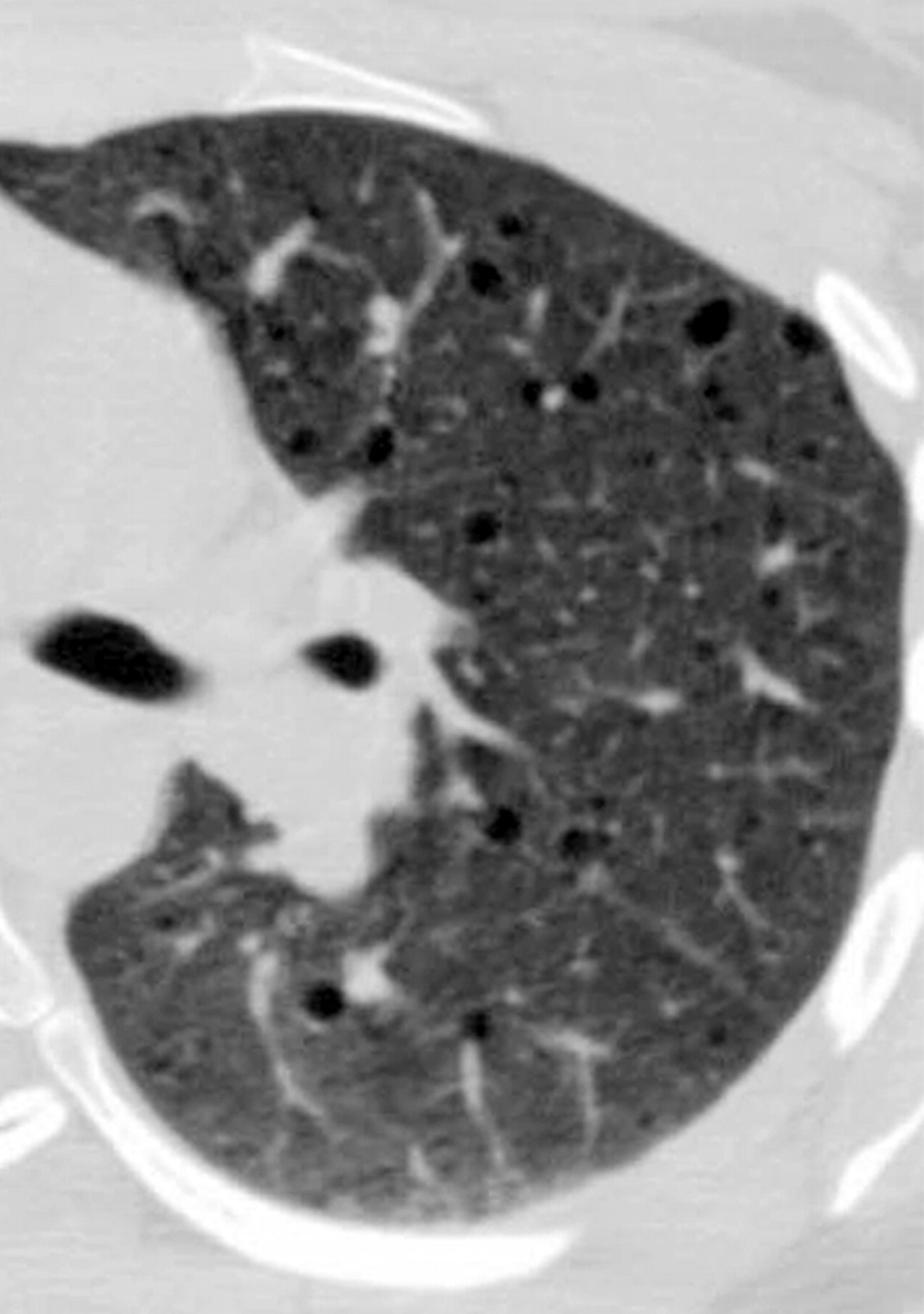
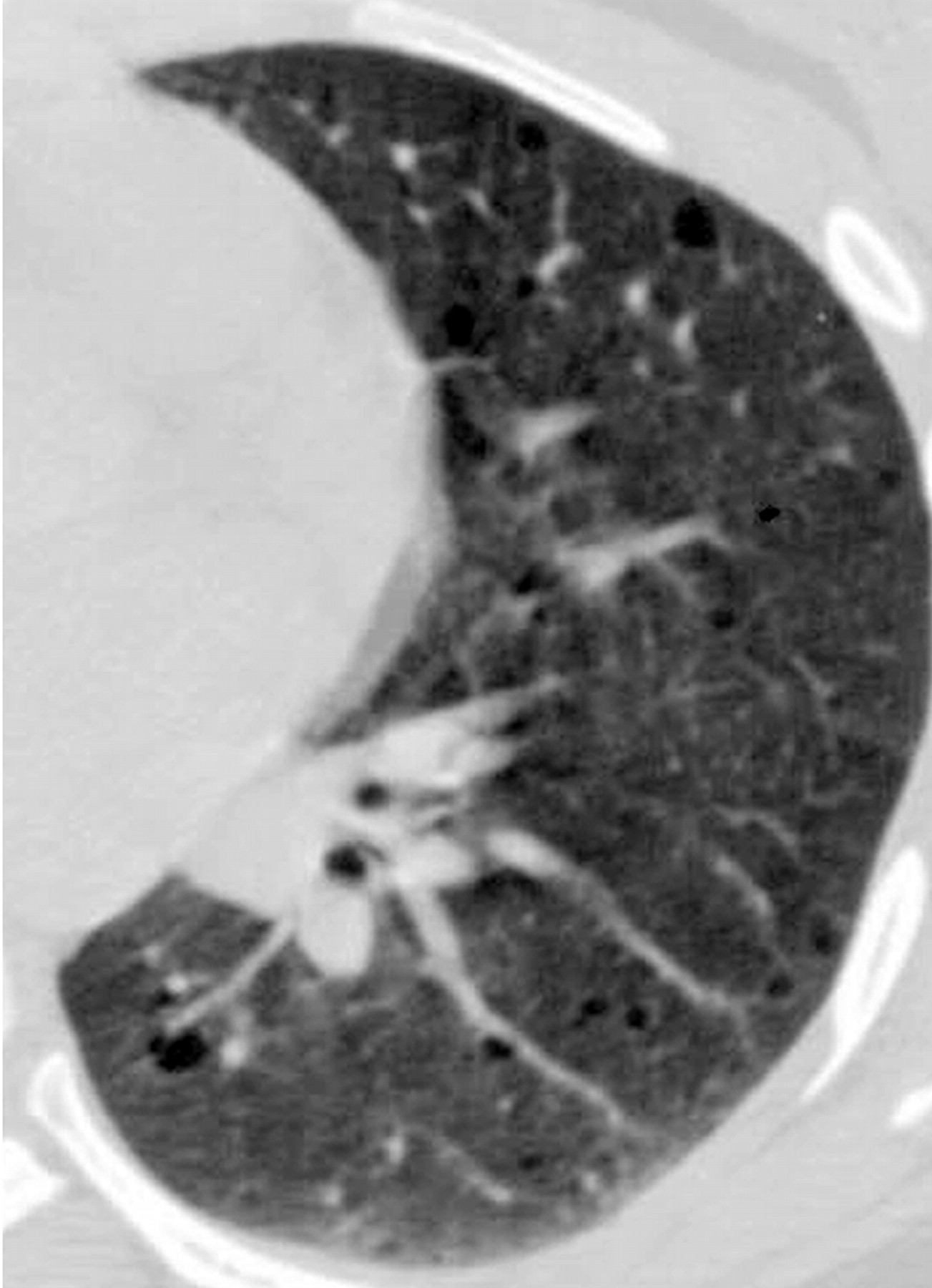
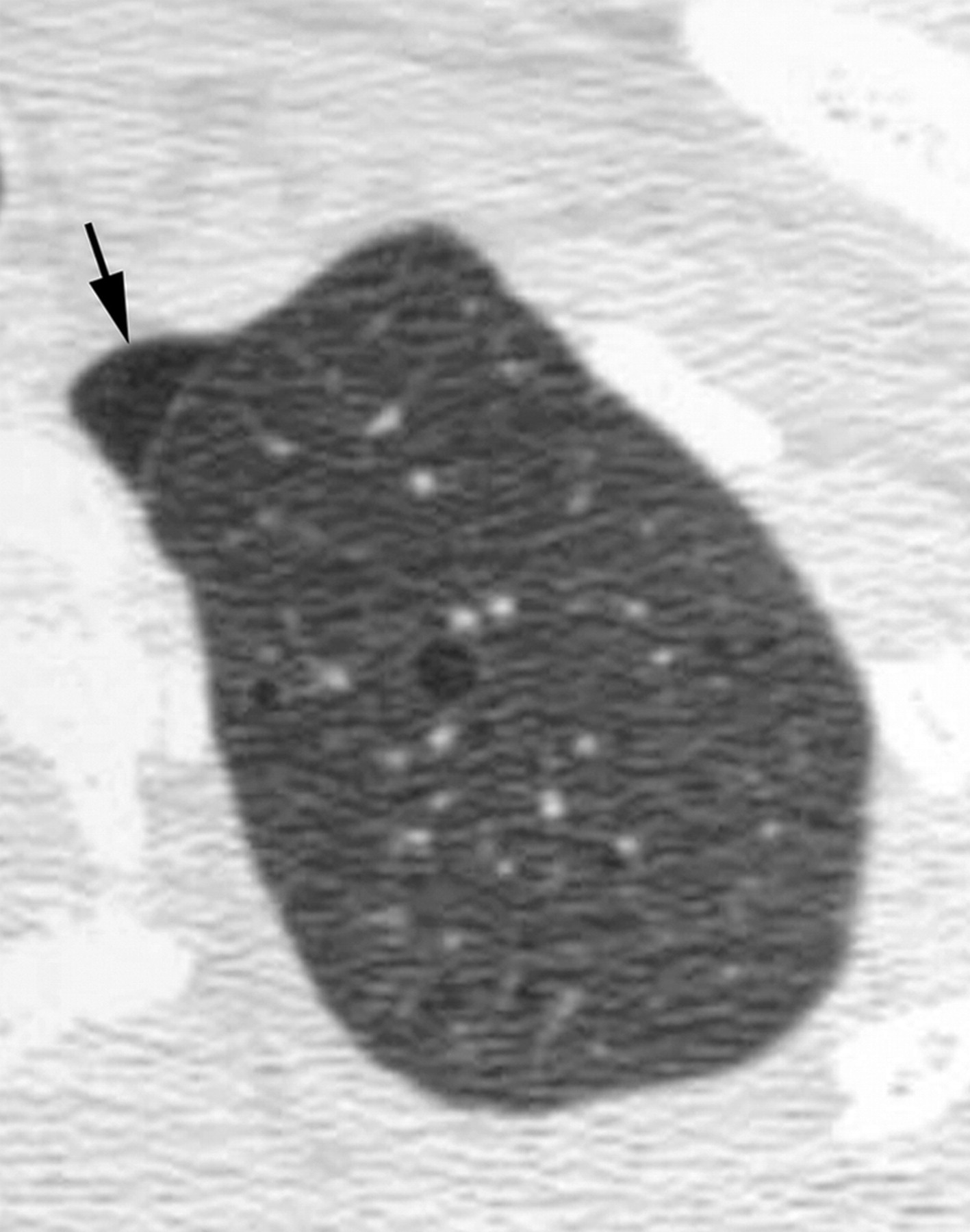
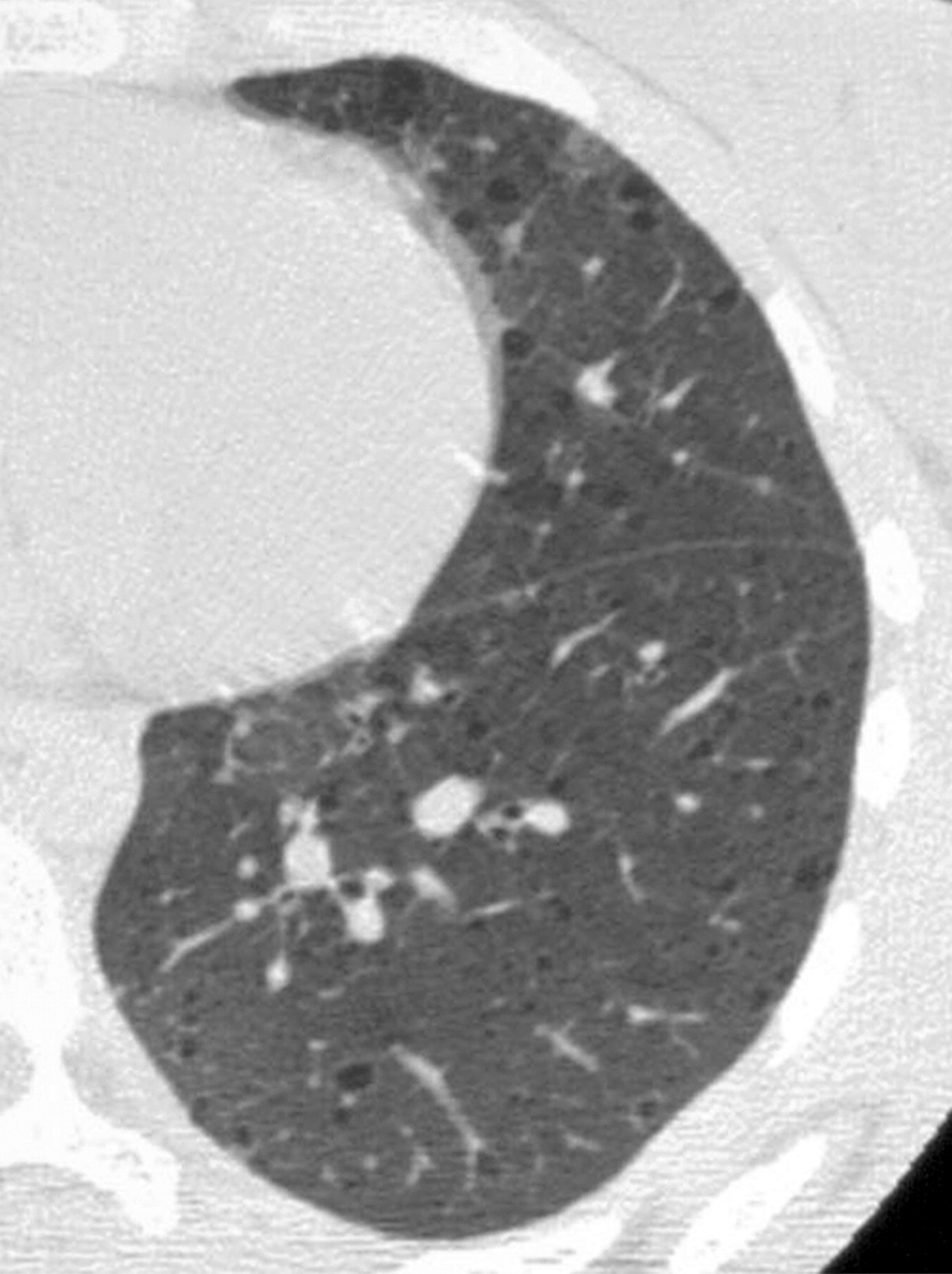
Наиболее характерный морфологический признак ЛАМ -кистозная трансформация легких, которая обычно выявляется на компьютерных томограммах. Кисты бывают двух типов: мелкие множественные типа «сотового легкого» и крупные кисты, присущие буллезной эмфиземе. Толщина стенки кисты не превышает 2 мм, причем стенка кисты выявляется не всегда и не на всем протяжении. Окружающая легочная ткань часто не изменена. Однако сочетание фиброзных и кистозных изменений не противоречит диагнозу ЛАМ. Таким образом, рентгенологическая картина ЛАМ не патогномонична. Ведущим рентгенологическим признаком этого заболевания является образование множественных воздушных тонкостенных полостей буллезного характера.
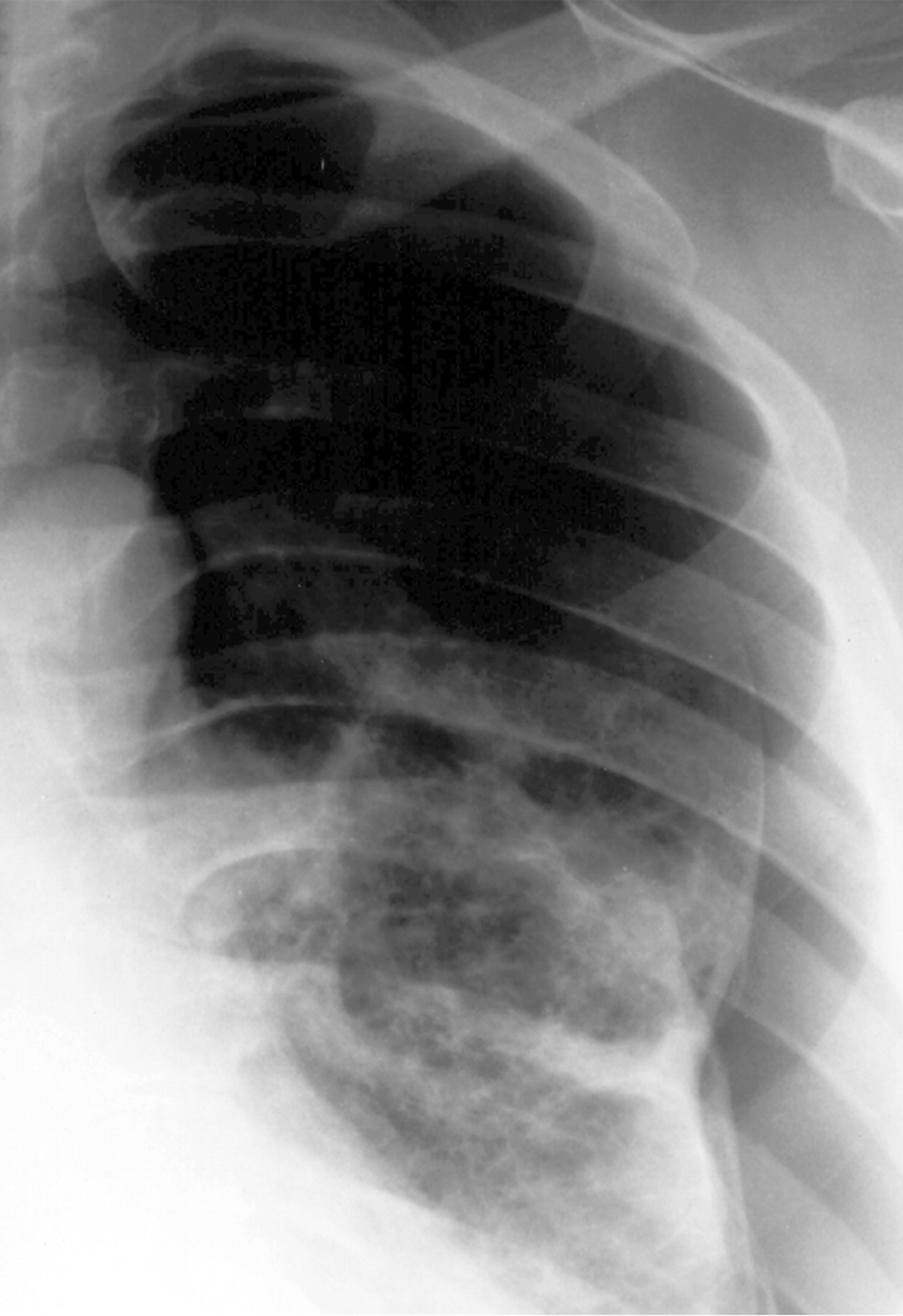
Для очаговой формы характерны очаги затемнения от 0,5 до 1,5 см в диаметре с четкими границами.
При развитии пневмоторакса определяется спавшееся поджатое воздухом легкое, при развитии хилоторакса - интенсивная гомогенная тень (за счет выпота) с косовосходящей верхней границей.
Компьютерная томография легких. Для ЛАМ характерны множественные диффузные, хорошо очерченные мелкие тонкостенные кисты. Кисты при этом заболевании значительно отличаются от зон центрилобулярной эмфиземы легких, которые не имеют четких границ и своих собственных стенок, а также от фибрози-рующего альвеолита, при котором основные изменения расположены по периферии легких, имеются поля фиброза и дезорганизации паренхимы легких, а кисты расположены субплеврально и характеризуются довольно толстыми стенками. Данные компьютерной томографии настолько специфичны для ЛАМ, что некоторые авторы для постановки точного диагноза полагают достаточным заключение компьютерной томографии (КТ) без проведения биопсии легких.
Некоторые исследователи показывают, что легочные кисты уменьшаются в объеме во время выдоха, что свидетельствует о наличии связи кист с воздухоносными путями. В связи с этим рекомендуется выполнять КТ во время глубокого вдоха и выдоха. Некоторые авторы считают, что изменения размеров воздушных образований в легких во время респираторной пробы характерны для кистозных бронхоэктазов, в отличие от субплевральных булл.
Многие авторы отмечают большие трудности дифференцирования ЛАМ с другими диффузными поражениями легких, в частности с гистиоцитозом Х, в связи с чем в целях окончательной диагностики считают необходимым производить биопсию легкого.
Исследование вентиляционной способности легких. Характерно увеличение остаточного объема легких в связи с образованием множественных кист. У большинства больных определяется также обструктивный тип дыхательной недостаточности: снижение объема форсированного выдоха за одну секунду (ОФВ1). Обструктивные изменения выявляют уже на самых ранних стадиях заболевания, поэтому больным может быть ошибочно установлен диагноз бронхиальной астмы или хронического обструктивного заболевания легких, даже при отсутствии соответствующей клинической картины. Однако для ЛАМ (в отличие от бронхиальной астмы или хронического обструктивного
заболевания легких) характерно выраженное снижение диффузионной способности легких. Рестриктивная дыхательная недостаточность (снижение ЖЕЛ) присоединяется по мере прогрес-сирования заболевания.
Исследование газов крови. По мере развития дыхательной недостаточности появляется артериальная гипоксемия, парциальное напряжение кислорода снижается, особенно после физической нагрузки.
Электрокардиография. По мере прогрессирования заболевания выявляются:
признаки гипертрофии миокарда правого предсердия и правого желудочка;
в отведениях II, III, aVF зубцы Р высокоамплитудные с заостренной вершиной (P-«pulmonale»);
в отведении V2 зубец Р (или, по крайней мере, его первая правопредсердная фаза) положительный с заостренной вершиной (P-«pulmonale»);
в отведениях I, aVL, V5, V6 зубец Р низкой амплитуды, а в аVL может быть отрицательным (непостоянный признак);
длительность зубца Р не превышает 0,10 с.;
смещение электрической оси сердца вправо (угол а более +100 градусов);
увеличение амплитуды зубца R в правых грудных отведениях (V1, V2) и амплитуды зубца S в левых грудных отведениях (V5, V6). При этом количественными критериями могут являться: амплитуда зубца RV1>7 мм или RV1+SV5, SV6>10,5 мм;
появление в отведении V1 комплекса QRS типа rSR‘ или QR;
признаки поворота сердца вокруг продольной оси по часовой стрелке (смещение переходной зоны влево, к отведениям V5-V6, и появление в отведениях V5, V6 комплекса QRS типа RS);
смещение сегмента RS-T вниз и появление отрицательных зубцов Т в отведениях III, aVF, V1, V2;
увеличение длительности интервала внутреннего отклонения в правом грудном отведении (V1) более 0,03 с.
Патоморфологическая диагностика
При патоморфологическом исследовании отмечаются следующие признаки заболевания:
значительное уплотнение ткани легких, множество мелких узелков 0,3-0,7 см в диаметре, белесоватых, заполненных жидкостью, они расположены субплеврально;
наличие в отдельных участках легких крупных воздушных полостей ;
гиперплазия лимфоузлов;
диффузная пролиферация гладкомышечных волокон в интерстиции легких (межальвеолярно, периваскулярно, перибронхиально, субплеврально, по ходу лимфатических сосудов);
деструктивные изменения стенок кровеносных и лимфатических сосудов, стенок бронхов, альвеол;
формирование микрокистозного «сотового» легкого;
развитие пневмо-гемо-хилоторакса в связи с деструкцией стенок кровеносных и лимфатических сосудов легких и развитием субплевральных кист.
При ЛАМ часто выявляют и внелегочные изменения: поражения медиастинальных и ретроперитонеальных лимфатических узлов, ангиомиолипомы (гамартомы).
Основывается на данных трансбронхиальной биопсии. Если трансбронхиальная биопсия не информативна, проводится открытая или торакоскопическая биопсия легких. Типичная морфологическая картина ЛАМ характеризуется пролиферацией гладкомышечных клеток в интерстиции и вокруг бронховаскуляр-ных структур. Пролиферирующие клетки напоминают миоциты сосудов, однако они более короткие, плейоморфные, и в ряде случаев их можно спутать с фиброцитами. В спорных случаях отличить атипичные гладкомышечные клетки при ЛАМ от других
Кафедра факультетской терапии №1 лечебного факультета Московского государственного медицинского университета им. И.М.Сеченова
С.И.Овчаренко, Е.А.Сон
Лимфангиолейомиоматоз легких (ЛАМ) – редкое заболевание, встречающееся только у женщин, проявляющееся кистозной перестройкой паренхимы легких с развитием прогрессирующей дыхательной недостаточности, а также узловой пролиферацией специфических ЛАМ-клеток, преимущественно в лимфатических сосудах и лимфатических узлах.
Впервые заболевание было описано в 1918 г. R. Lautenbacher [1]. Однако некоторые исследователи склонны отдавать пальму первенства E.von Stossel, описавшему это заболевание в 1937 г. у погибшей от дыхательной недостаточности женщины 43-летнего возраста [2]. К концу ХХ века было зафиксировано немногим более 100 случаев ЛАМ [3], причем в последние два десятилетия прошлого века за рубежом были описаны результаты наблюдений целых групп больных, включавших от 32 до 69 человек [4–6]. В нашей стране наибольшее количество случаев ЛАМ (23 случая) было зафиксировано к 2007 г. сотрудниками НИИ пульмонологии СПбГМУ им. акад. И.П.Павлова [7]. В 2006 г. в Регистре лимфангиолейомиоматоза Национального института сердца, легких и крови (The National Heart, Lung and Blood Institute – NHLBI) США было зарегистрировано уже 230 случаев этого заболевания [8], в том числе наблюдение пациентки, описанное нами в 2004 г. [9]. Таким образом, по самым обобщенным статистическим подсчетам, заболеваемость ЛАМ составляет 1–2 случая на 1 млн женщин [5, 6, 10], а ориентировочная численность больных может достигать 25–50 тыс. человек [11]. В настоящее время в связи с широким внедрением в клиническую практику компьютерной томографии (КТ) высокого разрешения (КТВР), позволяющей с высокой достоверностью выявлять характерные для ЛАМ изменения в легких, а также с появлением разработанных диагностических критериев болезни [12] это заболевание отмечают все чаще и чаще. Поэтому можно прогнозировать, что вследствие улучшения диагностики заболеваемость и распространенность ЛАМ в ближайшем будущем возрастет и практикующий врач будет верифицировать этот диагноз чаще. Несмотря на то что для ЛАМ характерно достаточно специфическое поражение легких, это заболевание имеет и системные проявления, в связи с чем ЛАМ попал как минимум в две различные классификации – классификацию гладкомышечных пролиферативных заболеваний и классификацию интерстициальных болезней легких. В 1983 г. E. Martin предложил классификацию множественных гладкомышечных поражений, основанную на соответствии клинической картины и рентгенологических изменений [13]. Автор выделил три основных варианта множественных лейомиоматозных поражений: 1. Метастатическая лейомиомиома (злокачественная лейомиосаркома). 2. Множественные фибролейомиоматозные гамартомы в легких. 3. Лейомиоматоз, в структуру которого и входит ЛАМ. В отличие от метастатической лейомиомы и множественных фибролейомиоматозных гамартом легких лейомиоматозом, а соответственно и ЛАМ, болеют только женщины. Лейомиоматоз включает в себя четыре нозологические формы: 1) лимфангиолейомиоматоз, для которого характерна прогрессирующая дыхательная недостаточность с формированием в финале заболевания «сотового легкого» и нередким сочетанием с хилотораксом, хилоасцитом и пневмотораксом; 2) доброкачественная метастазирующая лейомиома (узелковый лейомиоматоз, проявляющийся множественными узлами в легких); 3) лейомиоматозная диссеминация брюшины или перитонеальный лейомиоматоз; 4) внутривенный лейомиоматоз. В соответствие с классификацией 2000 г., разработанной совместно Американским торакальным и Европейским респираторным обществами (American Thoracic Sosiety/European Respiratory Sosiety – АТS/ERS), лимфангиолейомиоматоз включен в группу интерстициальных заболеваний легких, в структуре которых ЛАМ не относится ни к гранулематозам, ни к идиопатическим интерстициальным пневмониям, ни к интерстициальным болезням легких известной этиологии, а вместе с гистиоцитозом Х и прочими редкими заболеваниями составляет группу «другие». На сегодняшний день выделяют два варианта ЛАМ – спорадический и ассоциированный с туберозным склерозом (ТС) [12]. Спорадический ЛАМ протекает, как правило, тяжелее и быстрее приводит к формированию дыхательной недостаточности и инвалидизации больного. В связи с этим при верификации ЛАМ принципиально важно своевременно выявить имеющийся у пациента ТС, поскольку данный факт имеет большое прогностическое значение и определяет тактику ведения пациента. ТС, или болезнь Бурневилля–Прингла, – системная наследственная дисплазия, обусловленная нарушением закладки эктодермального зародышевого листка, которая характеризуется комбинированным опухолевидным поражением кожи, головного мозга, глазных яблок, сердца, почек и легких [14]. ТС – аутосомно-доминантное заболевание c неполной пенетрантностью, обусловленное различными мутациями генов TSC1 и/или TSC2 (Tuberous Sclerosis Complex). В соответствии с существующей на сегодняшний день классификацией наследственных дисплазий ТС относится к факоматозам (phakos – от греч. чечевица, родимое пятно) – нейроэктодермальным заболеваниям, включающим кроме ТС такие редкие нозологические формы, как нейрофиброматоз, синдром Стерджа–Вебера и болезнь Гиппеля–Линдау. ТС верифицируется в соответствие с критериями диагностики этого заболевания, которые подразделяются на «большие» и «малые» [15]. Большие диагностические критерии ТС К большим критериям диагноза ТС относятся: ангиофиброматоз лица (щеки, спинка носа); подногтевые фибромы; три пятна гипопигментации и более, полиоз; участки в виде шагреневых бляшек; множественные гамартомные узелки на сетчатке; бугорки в коре больших полушарий; субэпендимальные узелки; субэпендимальная гигантоклеточная астроцитома; рабдомиома сердца; почечные ангиомиолипомы или ЛАМ. Малые диагностические критерии ТС Среди малых диагностических критериев ТС можно назвать множественные ямки на эмали зубов; гамартомные полипы прямой кишки; костные кисты; фиброматоз десен; непочечные гамартомы; ахроматические пятна на сетчатке; кожные проявления в виде конфетти (мелкие круглые пятна); множественные кисты почек; миграция белого вещества мозга в виде линий луча. Диагноз ТС считается неоспоримым при наличии 2 больших критериев или 1 большого и 2 малых. Вероятный диагноз ставится при наличии 1 большого и 1 малого критерия. Диагноз ТС считается сомнительным при наличии 1 большого критерия. В отличие от ЛАМ, которым страдают преимущественно женщины, ТС встречается с одинаковой частотой у лиц обоих полов. Распространенность ТС значительно выше, чем распространенность ЛАМ, и в случае диагностики в детском возрасте варьирует от 1 на 6800 до 1 на 17 300 [15]. Примерно 1/3 взрослых пациентов с ТС имеют рентгенологические признаки ЛАМ [16, 17]. Столь частая распространенность ЛАМ среди лиц, страдающих ТС, естественно, наводит на мысль об этиологическом и/или патогенетическом родстве этих заболеваний. Этиология ЛАМ пока до конца не изучена, однако установлено, что пациенты, страдающие ЛАМ, имеют мутации в тех же генах, что и лица с ТС, – в генах TSC1 и TSC2 [18]. Следует отметить, что мутации в генах ТС обнаруживаются не только при ЛАМ, ассоциированном с ТС, но и при спорадическом его варианте, т.е. при отсутствии болезни Бурневилля–Прингла. Наличие генных мутаций, безусловно, свидетельствует в пользу генетической детерминированности ЛАМ. Однако в отличие от ТС, передающегося по наследству, передача ЛАМ от матери к дочери не описана [11]. Все эти факты позволяют сделать вывод о том, что обе эти нозологические формы являются генетическими заболеваниями, поскольку обусловлены мутациями в одних и тех же генах ТС (TSC1 и TSC2), но при ТС мутации в этих генах являются гаметическими, т.е. возникают в гаметах и, следовательно, передаются по наследству, а при ЛАМ мутации в тех же генах являются соматическими (т.е. возникают не в гаметах, а в клетках тканей организма), вследствие чего и не передаются последующим поколениям. Таким образом, хотя ЛАМ, как и ТС, является генетической нозологией, в отличие от последнего он не является наследственным заболеванием. Ген TSC1 кодирует синтез белка гамартина, а ген TSC2 – белка туберина. Гамартин и туберин образуют гамартин-тубериновый комплекс, который является ключевым посредником в суммировании информации о ростовой стимуляции и подавляет чрезмерный клеточный рост через mTOR (мишени для иммуносупрессанта рапамицина у млекопитающих) [15]. При ТС возникают инактивирующие мутации в обоих генах-супрессорах – TSC1 и/или TSC2, что приводит к снижению активности гамартин-туберинового комплекса, угнетающего избыточный клеточный рост, а следовательно, препятствующего росту гамартом. Для ЛАМ более характерны мутации в гене TSC2 [19]. Наличие дефекта гена TSC2 предположительно является причиной аномального ответа (роста) ЛАМ-клеток (атипичных гладкомышечных и эпителиоидных) на женские половые гормоны [15]. Эстрогены регулируют транскрипцию многих генов, кроме этого, они могут играть роль стимула в пролиферации и миграции гладкомышечных клеток в другие органы и ткани. У больных ЛАМ выявляются как точечные мутации гена TSC2, так и мутации, связанные с утратой гетерозиготности локуса TSC2, приводящие к потере функции подавления опухолевого роста и, следовательно, бесконтрольной пролиферации клеток («двойной удар по генам-супрессорам опухолей»). Несмотря на выявленные мутации в генах TSC1 и TSC2, патогенез ЛАМ до конца не известен. Суммируя накопленные на сегодняшний день данные об этом заболевании, с патогенетической точки зрения ЛАМ можно рассматривать как многофокусный гамартомный (опухолеподобный) процесс, обусловленный генетическим дефектом, который определяет аномальный ответ мышечных клеток на женские половые гормоны, возможно, играющие важную роль в прогрессировании заболевания. В действительности об опухолеподобном характере ЛАМ свидетельствуют некоторые характерные для злокачественных новообразований черты, присущие этому заболеванию. Классическими признаками злокачественности служат: 1) автономно пролиферирующий бессмертный клон клеток, непрерывно эволюционирующий в сторону независимости от контроля организма; 2) инвазия и 3) метастазирование [20]. 1. Морфологический субстрат ЛАМ – это до некоторой степени бесконтрольный опухолевоподобный рост ЛАМ-клеток в интерстиции легких, лимфатических сосудах и узлах [21]. ЛАМ-клетки представляют собой неупорядоченно пролиферирующие клетки двух типов – эпителиоидные (внешне напоминающие эпителиальные) и гладкомышечные клетки. Видовая принадлежность этих клеток пока не установлена, поскольку, с одной стороны, они дают, как и полагается гладкомышечным клеткам, положительную реакцию на актин, виментин и десмин, а с другой – экспрессируют белок премеланоцита. Отсутствие фигур митоза исключает подозрение на злокачественный характер этой пролиферации [22]. 2. В пользу «инвазивности» роста ЛАМ-клеток свидетельствует повышенное содержание в них SRF (serum response factor). SRF – сывороточный ответный фактор, увеличивающий экспрессию матриксных металлопротеиназ (ММП) 2-го (коллагеназа IV типа) и 14-го (мембраносвязанная ММП) типов, содержание которых при ЛАМ повышается. Как известно, степень инвазивного роста и метастазирования опухолевых клеток определяется их способностью расщеплять компоненты экстрацеллюлярного матрикса, такие как базальные мембраны и компоненты межтканевой стромы (коллаген, эластин, ламинин и др.), что достигается с помощью повышенного содержания ММП [23]. В отличие от многих протеолитических ферментов, способных расщеплять отдельные компоненты экстрацеллюлярного матрикса, ММП разрушают все его структуры, что и позволяет опухолевым клеткам внедряться на территорию других тканей. Следует добавить, что наряду с увеличением экспрессии ММП при ЛАМ отмечается снижение уровня их тканевых ингибиторов 3-го типа (TIMP-3). Однако следует отметить, что повышение уровня ММП также характерно и для других интерстициальных заболеваний легких, таких как идиопатический легочный фиброз и саркоидоз [24]. 3. Говоря о ЛАМ как о гамартомном (опухолеподобном) заболевании, способном к метастазированию, можно упомянуть о возникновении ЛАМ в донорском легком после трансплантации [25, 26]. Некоторые исследователи рассматривают поражение легких при ЛАМ как следствие метастазирования из других органов, в которых выявляются сходные изменения (почки, лимфатические узлы) [19, 27]. К фактам, свидетельствующим о важной роли эндокринных воздействий, в первую очередь женских половых гормонов, можно отнести возникновение ЛАМ у женщин, преимущественно детородного возраста; дебют и обострение ЛАМ могут быть спровоцированы лечением эстрогенами; во время менструации или беременности наблюдается клиническое ухудшение, проявляющееся в нарастании одышки и развитии пневмотораксов, нередко рецидивирующих; после наступления менопаузы или удаления яичников прогрессирование заболевания в ряде случаев замедляется. ЛАМ часто сочетается с фибро- или лейомиомой матки [28–30]. В 80% случаев на ЛАМ-клетках обнаруживаются поверхностные PRs- (прогестероновые) и ERs- (эстрогеновые) рецепторы, что подтверждает гормональную зависимость заболевания [7]. Резюмируя сказанное, можно говорить о ведущей роли популяции ЛАМ-клеток в патогенезе заболевания. ЛАМ-клетки обладают фенотипом, способным к бесконтрольному росту, экспрессии гормональных рецепторов и протеаз, а также склонным к метастатическому поведению. Пролиферация ЛАМ-клеток преимущественно имеет место в легких и в лимфатической системе, что и определяет клинические проявления этого заболевания, которые можно разделить на легочные и внелегочные соответственно. В легких пролиферация ЛАМ-клеток выявляется вокруг бронхиол, артерий, вен, в лимфатических сосудах, а также в межальвеолярных перегородках и плевре [22, 31]. Сужение просвета мелких бронхиол, вероятно, за счет формирования «воздушных ловушек» приводит к гиперинфляции с последующим образованием мелких кист, что и объясняет макроскопическую картину микрокистозной перестройки паренхимы. При разрыве кист, расположенных подплеврально, развиваются пневмотораксы, часто рецидивирующие. В финале такой перестройки легочной паренхимы развивается дыхательная недостаточность, клинически проявляющаяся прогрессирующей одышкой. Разрастание ЛАМ-клеток в сосудах малого круга также играет значимую роль в патогенезе и клинической картине этого заболевания. Сужение просвета артериол приводит к значительному росту сосудистого сопротивления в малом круге кровообращения и, следовательно, к легочной гипертензии с последующим закономерным формированием легочного сердца. Разрастание ЛАМ-клеток вокруг венул приводит к их компрессии с сужением просвета и повышением давления внутри сосудов, к нарушению целостности сосудистой стенки, геморрагиям, гемосидерозу, а клинически проявляется кровохарканьем [21, 32]. При сдавлении лимфатических сосудов легких и плевры узелками пролиферирующих ЛАМ-клеток с их последующей обтурацией и разрывами развиваются хилоторакс и хилоптоэ. Хилоторакс имеет место у 28% больных ЛАМ [4], в то время как в общей структуре плевральных выпотов по частоте встречаемости он занимает одно из последних мест. При подобном поражении лимфатических сосудов брюшной полости, таза и ретроперитонеального пространства развивается хилезный асцит. Специфическое для ЛАМ поражение лимфатической системы – развитие лимфангиолейомиом – скопление увеличенных и кистозно-расширенных лимфатических сосудов, за счет инфильтрации ЛАМ-клетками. Поражение лимфатической системы при ЛАМ может проявляться лимфаденопатией. Кроме того, внелегочными проявлениями ЛАМ являются ангиомиолипомы почек и менингиомы. При анализе симптомов, которые встречаются при ЛАМ, можно говорить о том, что в клинической картине этого заболевания доминирующей является прогрессирующая одышка с постепенным снижением толерантности к физической нагрузке, встречающаяся в 94% случаев [4] и являющаяся проявлением дыхательной недостаточности. Спонтанный пневмоторакс развивается в 80–81% случаев [4, 33] и может быть первым проявлением заболевания. У 41% больных были жалобы на кашель, в основном непродуктивного характера. Кровохарканье и боли в грудной клетке предъявляли 44 и 34% пациентов соответственно, а у 28% пациентов был обнаружен хилоторакс [4]. Кроме этого, описано развитие стридорозного дыхания и такие внелегочные поражения, как ангиомиолипомы почек. Возможно бессимптомное течение заболевания. Диагноз ЛАМ устанавливают в соответствии с разработанными критериями и может быть оценен как достоверный, вероятный или сомнительный [12]. Диагностические критерии лимфангиолейомиоматоза Достоверный диагноз ЛАМ может быть поставлен, если выявляются: 1. «Характерные для ЛАМ легких» или «совместимые с ЛАМ легких» КТ-признаки и биопсия легких, соответствующая морфологическим критериям для ЛАМ или 2. «Характерные для ЛАМ легких» КТ-признаки и любой из следующих: • ангиомиолипома (почки); • хилезный выпот в грудной или брюшной полости; • лимфангиолейомиома или поражение лимфоузлов, характерное для ЛАМ; • определенный или возможный ТС. Вероятный диагноз ЛАМ верифицируется, если имеются следующие проявления: 1. «Характерные для ЛАМ легких» КТ-признаки и клиническая картина или 2. «Совместимые с ЛАМ легких» КТ-признаки и любой из следующих: ангиомиолипома почки, хилезный выпот в грудной или брюшной полости. Сомнительный диагноз ЛАМ базируется лишь на КТ-признаках: 1. «Характерные для ЛАМ легких» или «совместимые с ЛАМ легких» КТ-признаки. По степени специфичности для ЛАМ рентгенологические критерии подразделены на «характерные для ЛАМ легких» и «совместимые с ЛАМ легких». Характерные для ЛАМ легких КТ-признаки являются более специфичными для ЛАМ и включают множественные тонкостенные круглые хорошо дифференцируемые воздушные кисты при сохранном или увеличенном объеме легких без других значимых поражений, отсутствие признаков других интерстициальных болезней легких, за исключением маленьких узелков, которые наиболее часто служат КТ-проявлением мультифокальной микроузловой гиперплазии пневмоцитов у пациентов с ТС (как без, так и в сочетании с ЛАМ). К совместимым с ЛАМ легких изменениям, выявляемым при КТВР, относят наличие только небольшого количества (от 2 до 10) таких кист, размером до 30 мм в диаметре, при соблюдении прочих перечисленных выше условий. К наиболее информативным диагностическим методам при ЛАМ относят КТВР и морфологическое исследование ткани легкого, поскольку для этих методов разработаны наиболее достоверные критерии. Сочетание положительных результатов обоих этих исследований позволяют убедительно диагностировать ЛАМ [12]. По данным большинства имеющихся на сегодняшний день исследований, к «золотому стандарту» диагностики относят морфологическое исследование биоптата ткани легкого. Критериями диагноза, на основании наличия которых опытный патологоанатом может диагностировать ЛАМ, служат одновременное присутствие множественных мелких воздушных кист и наличие мультифокальной узловой пролиферации двух основных видов ЛАМ-клеток – незрелых гладкомышечных и периваскулярных эпителиоидных клеток. Поскольку опыт морфолога в диагностике столь редкого заболевания не всегда позволяет сделать однозначные выводы, а также в случаях, когда морфологическая картина, особенно на ранних стадиях заболевания, не вполне специфична, целесообразно проведение дополнительного иммуногистохимического исследования для выявления экспрессии α-гладкомышечного актина и НМВ-45. Кроме этого, полезным дополнением при постановке морфологического диагноза может быть определение рецепторов к эстрогену и прогестерону. Тем не менее в соответствии с разработанными на сегодняшний день критериями диагностики ЛАМ достоверный диагноз может быть верифицирован и без проведения исследования биоптата [12, 13]. Распространение в последние годы в клинической практике КТВР позволило выявить рентгенологические маркеры поражения легких при ЛАМ, что сделало этот метод исследования обязательным в диагностике данного заболевания, поскольку на основании убедительных КТ-признаков в сочетании с соответствующей клинической картиной ЛАМ можно диагностировать с высокой достоверностью и без проведения биопсии легкого [12]. В тех случаях, когда достоверный диагноз ЛАМ ставится без результатов биопсии легких, помимо результатов КТВР большое значение придается как легочным, так и внелегочным клиническим проявлениям заболевания. Как указывалось выше, внелегочными критериями диагностики ЛАМ являются наличие ангиомиолипомы почки (подтверждается при наличии КТ- и/или морфологических критериев опухоли), хилезного выпота в плевральной или брюшной полости (характер выпота устанавливается либо визуально, либо по биохимическим маркерам), лимфангиолейомиомы или лимфаденопатии, характерной для ЛАМ (с обязательной морфологической верификацией) или достоверного или вероятного диагноза ТС, который устанавливается в соответствии с критериями диагноза болезни Бурневилля–Прингла [15]. В соответствии с разработанными критериями диагностики вероятный диагноз ЛАМ может быть поставлен при сочетании характерных КТ-признаков с клинической картиной, включающей множественный и/или билатеральный пневмоторакс и/или соответствующие ЛАМ изменения функции легких, которые состоят в преимущественном снижении TLCO и в меньшей степени объема форсированного выдоха за 1-ю секунду при сохранных легочных объемах [12, 13]. На сегодняшний день эффективного лечения ЛАМ не существует. Пациентам главным образом проводится симптоматическая терапия: бронходилататоры, кислородотерапия. Предпринимаются попытки патогенетической терапии, основанные на данных о неблагоприятном влиянии эстрогенов на течение заболевания: антиэстрогенные препараты (тамоксифен, прогестерон) и редукция синтеза эстрогенов (оофорэктомия, радиоабляция яичников). Убедительных данных об эффективности этих методов лечения не получено, но наиболее целесообразным считается отмена эстрогенных препаратов, в том случае, если лечение ими проводится в связи с другими заболеваниями, а также предохранение от беременности. Вопрос о трансплантации легких, принимая во внимание возможность рецидива в трансплантате, также пока остается весьма дискутабельным. На сегодняшний день показаниями для трансплантации являются III или IV функциональный класс по NYHA с гипоксемией в покое, тяжелым нарушением легочной функции и дееспособности [12].






Продолжение.
Лимфангиомиоматоз.
Лимфангиолейомиоматоз: современный взгляд на проблему
Н.И. ШВЕЦ, д. мед. н., профессор; Т.М. БЕНЦА, к. мед. н., доцент; В.В. СТАНИШЕВСКИЙ
/Национальная медицинская академия последипломного образования им. П.Л. Шупика, Киев/
Лимфангиолейомиоматоз (лейомиоматоз) (ЛАМ) - это редкая патология, возникающая у женщин детородного возраста 18-50 лет; характеризуется прогрессирующей одышкой, пневмо-, хилотораксом и кровохарканьем. В основе ЛАМ - диссеминиро-ванный патологический процесс, характеризующийся опухолевидным разрастанием гладкомышечных волокон по ходу мелких бронхов, бронхиол, стенок кровеносных и лимфатических сосудов легких с последующей мелкокистозной трансформацией легочной ткани. Основное проявление ЛАМ - прогрессирующая дыхательная недостаточность.
Диффузный ЛАМ (диффузный лейомиоматоз легких, легочной лейомиоматоз, фибролейомиоматозная гамартома) принадлежит к числу редко встречающихся заболеваний. Первое описание ЛАМ датировано 1937 годом. С этого времени зарегистрировано немногим более 100 случаев ЛАМ. Однако за последние 5 лет отмечается резкий рост данной патологии в странах Европы.
Этиология и патогенез
Этиология ЛАМ остается неизвестной. Предполагают гормо-нозависимость (эстрогенозависимость) заболевания. Косвенно эта версия подтверждается тем, что ЛАМ встречается преимущественно у женщин репродуктивного возраста, крайне редко -у мужчин. Обостряется заболевание во время беременности, в предменструальном периоде, а стабилизация процесса отмечается в постменопаузе. Сочетание ЛАМ легких с лейомиомой матки также указывает на важную роль эндокринных нарушений в развитии болезни. Не исключено, что возникновение заболевания связано с иммунными нарушениями. Имеются также данные о том, что определенную роль в развитии ЛАМ играют генетические нарушения в белках, вовлеченных в синтез катехоламинов.
Существующие теории возникновения заболевания не объясняют в полной мере его причину. Наибольшее клиническое подтверждение находит теория гормональных нарушений. По другим данным, в основе заболевания лежит асинхронная мышечная пролиферация в легких, матке и, возможно, в мышцах другой локализации. Еще одна теория основана на том, что лейомиома-тозные узлы возникают через 1-20 лет после удаления матки по поводу фибромиомы, что связано с эмболией сосудистого русла гладкомышечными клетками.
Клиника
В начальной стадии клинические проявления могут отсутствовать. Длительное время заболевание протекает бессимптомно. ЛАМ часто обнаруживается случайно как диффузное или мелкоузловое поражение легочной ткани при рентгенологическом исследовании органов грудной клетки. Пациенты обращаются за помощью при появлении прогрессирующей одышки вследствие развивающейся обструкции дыхательных путей и снижения диффузионной способности легочной ткани.
Основные клинические проявления:
Очаговая форма ЛАМ протекает бессимптомно и выявляется рентгенологически. В некоторых случаях заболевание принимает системный характер - лейомиомы развиваются в брюшной полости, забрюшинном пространстве, матке, кишечнике, почках. Ангиомиолипомы почек редко нарушают функцию почек, хотя иногда могут достигать больших размеров (более 10 см).
Активации заболевания способствуют беременность, роды, прием контрацептивов. Прогноз у таких больных, как правило, неблагоприятный. Летальный исход наступает в сроки от двух до 10-ти лет. Средняя продолжительность жизни больных составляет около 5 лет. Описаны случаи с летальным исходом через 17 лет. Непосредственная причина смерти - прогрессирующая дыхательная недостаточность.
Лабораторные методы диагностики
Общий анализ крови: существенных изменений нет. У некоторых больных отмечается эозинофилия, нередко увеличивается СОЭ, особенно при развитии пневмо-хилоторакса.
Общий анализ мочи: может наблюдаться незначительная протеинурия (симптом неспецифический и непостоянный).
Биохимическое исследование крови: иногда наблюдается гиперхолестеринемия, возможно увеличение уровня а2- и у-глобулинов, аминотрансфераз, общей лактатдегидрогеназы, ангиотензинпревращающего фермента
Исследование плевральной жидкости: хилоторакс чрезвычайно характерен для ЛАМ. Плевральная жидкость имеет следующие характерные особенности:
Инструментальные методы исследования
Рентгенологическое исследование легких. Главными рентгенологическими признаками ЛАМ легких на обычных рентгенограммах грудной клетки являются:
Наиболее характерный морфологический признак ЛАМ -кистозная трансформация легких, которая обычно выявляется на компьютерных томограммах. Кисты бывают двух типов: мелкие множественные типа «сотового легкого» и крупные кисты, присущие буллезной эмфиземе. Толщина стенки кисты не превышает 2 мм, причем стенка кисты выявляется не всегда и не на всем протяжении. Окружающая легочная ткань часто не изменена. Однако сочетание фиброзных и кистозных изменений не противоречит диагнозу ЛАМ. Таким образом, рентгенологическая картина ЛАМ не патогномонична. Ведущим рентгенологическим признаком этого заболевания является образование множественных воздушных тонкостенных полостей буллезного характера.
Для очаговой формы характерны очаги затемнения от 0,5 до 1,5 см в диаметре с четкими границами.
При развитии пневмоторакса определяется спавшееся поджатое воздухом легкое, при развитии хилоторакса - интенсивная гомогенная тень (за счет выпота) с косовосходящей верхней границей.
Компьютерная томография легких. Для ЛАМ характерны множественные диффузные, хорошо очерченные мелкие тонкостенные кисты. Кисты при этом заболевании значительно отличаются от зон центрилобулярной эмфиземы легких, которые не имеют четких границ и своих собственных стенок, а также от фибрози-рующего альвеолита, при котором основные изменения расположены по периферии легких, имеются поля фиброза и дезорганизации паренхимы легких, а кисты расположены субплеврально и характеризуются довольно толстыми стенками. Данные компьютерной томографии настолько специфичны для ЛАМ, что некоторые авторы для постановки точного диагноза полагают достаточным заключение компьютерной томографии (КТ) без проведения биопсии легких.
Некоторые исследователи показывают, что легочные кисты уменьшаются в объеме во время выдоха, что свидетельствует о наличии связи кист с воздухоносными путями. В связи с этим рекомендуется выполнять КТ во время глубокого вдоха и выдоха. Некоторые авторы считают, что изменения размеров воздушных образований в легких во время респираторной пробы характерны для кистозных бронхоэктазов, в отличие от субплевральных булл.
Многие авторы отмечают большие трудности дифференцирования ЛАМ с другими диффузными поражениями легких, в частности с гистиоцитозом Х, в связи с чем в целях окончательной диагностики считают необходимым производить биопсию легкого.
Исследование вентиляционной способности легких. Характерно увеличение остаточного объема легких в связи с образованием множественных кист. У большинства больных определяется также обструктивный тип дыхательной недостаточности: снижение объема форсированного выдоха за одну секунду (ОФВ1). Обструктивные изменения выявляют уже на самых ранних стадиях заболевания, поэтому больным может быть ошибочно установлен диагноз бронхиальной астмы или хронического обструктивного заболевания легких, даже при отсутствии соответствующей клинической картины. Однако для ЛАМ (в отличие от бронхиальной астмы или хронического обструктивного
заболевания легких) характерно выраженное снижение диффузионной способности легких. Рестриктивная дыхательная недостаточность (снижение ЖЕЛ) присоединяется по мере прогрес-сирования заболевания.
Исследование газов крови. По мере развития дыхательной недостаточности появляется артериальная гипоксемия, парциальное напряжение кислорода снижается, особенно после физической нагрузки.
Электрокардиография. По мере прогрессирования заболевания выявляются:
Патоморфологическая диагностика
При патоморфологическом исследовании отмечаются следующие признаки заболевания:
При ЛАМ часто выявляют и внелегочные изменения: поражения медиастинальных и ретроперитонеальных лимфатических узлов, ангиомиолипомы (гамартомы).
Гистологическое подтверждение диагноза лимфангиолейомиоматоза
Основывается на данных трансбронхиальной биопсии. Если трансбронхиальная биопсия не информативна, проводится открытая или торакоскопическая биопсия легких. Типичная морфологическая картина ЛАМ характеризуется пролиферацией гладкомышечных клеток в интерстиции и вокруг бронховаскуляр-ных структур. Пролиферирующие клетки напоминают миоциты сосудов, однако они более короткие, плейоморфные, и в ряде случаев их можно спутать с фиброцитами. В спорных случаях отличить атипичные гладкомышечные клетки при ЛАМ от других
Впервые заболевание было описано в 1918 г. R. Lautenbacher [1]. Однако некоторые исследователи склонны отдавать пальму первенства E.von Stossel, описавшему это заболевание в 1937 г. у погибшей от дыхательной недостаточности женщины 43-летнего возраста [2]. К концу ХХ века было зафиксировано немногим более 100 случаев ЛАМ [3], причем в последние два десятилетия прошлого века за рубежом были описаны результаты наблюдений целых групп больных, включавших от 32 до 69 человек [4–6]. В нашей стране наибольшее количество случаев ЛАМ (23 случая) было зафиксировано к 2007 г. сотрудниками НИИ пульмонологии СПбГМУ им. акад. И.П.Павлова [7]. В 2006 г. в Регистре лимфангиолейомиоматоза Национального института сердца, легких и крови (The National Heart, Lung and Blood Institute – NHLBI) США было зарегистрировано уже 230 случаев этого заболевания [8], в том числе наблюдение пациентки, описанное нами в 2004 г. [9]. Таким образом, по самым обобщенным статистическим подсчетам, заболеваемость ЛАМ составляет 1–2 случая на 1 млн женщин [5, 6, 10], а ориентировочная численность больных может достигать 25–50 тыс. человек [11]. В настоящее время в связи с широким внедрением в клиническую практику компьютерной томографии (КТ) высокого разрешения (КТВР), позволяющей с высокой достоверностью выявлять характерные для ЛАМ изменения в легких, а также с появлением разработанных диагностических критериев болезни [12] это заболевание отмечают все чаще и чаще. Поэтому можно прогнозировать, что вследствие улучшения диагностики заболеваемость и распространенность ЛАМ в ближайшем будущем возрастет и практикующий врач будет верифицировать этот диагноз чаще.
Несмотря на то что для ЛАМ характерно достаточно специфическое поражение легких, это заболевание имеет и системные проявления, в связи с чем ЛАМ попал как минимум в две различные классификации – классификацию гладкомышечных пролиферативных заболеваний и классификацию интерстициальных болезней легких.
В 1983 г. E. Martin предложил классификацию множественных гладкомышечных поражений, основанную на соответствии клинической картины и рентгенологических изменений [13]. Автор выделил три основных варианта множественных лейомиоматозных поражений:
1. Метастатическая лейомиомиома (злокачественная лейомиосаркома).
2. Множественные фибролейомиоматозные гамартомы в легких.
3. Лейомиоматоз, в структуру которого и входит ЛАМ.
В отличие от метастатической лейомиомы и множественных фибролейомиоматозных гамартом легких лейомиоматозом, а соответственно и ЛАМ, болеют только женщины. Лейомиоматоз включает в себя четыре нозологические формы:
1) лимфангиолейомиоматоз, для которого характерна прогрессирующая дыхательная недостаточность с формированием в финале заболевания «сотового легкого» и нередким сочетанием с хилотораксом, хилоасцитом и пневмотораксом;
2) доброкачественная метастазирующая лейомиома (узелковый лейомиоматоз, проявляющийся множественными узлами в легких);
3) лейомиоматозная диссеминация брюшины или перитонеальный лейомиоматоз;
4) внутривенный лейомиоматоз.
В соответствие с классификацией 2000 г., разработанной совместно Американским торакальным и Европейским респираторным обществами (American Thoracic Sosiety/European Respiratory Sosiety – АТS/ERS), лимфангиолейомиоматоз включен в группу интерстициальных заболеваний легких, в структуре которых ЛАМ не относится ни к гранулематозам, ни к идиопатическим интерстициальным пневмониям, ни к интерстициальным болезням легких известной этиологии, а вместе с гистиоцитозом Х и прочими редкими заболеваниями составляет группу «другие».
На сегодняшний день выделяют два варианта ЛАМ – спорадический и ассоциированный с туберозным склерозом (ТС) [12]. Спорадический ЛАМ протекает, как правило, тяжелее и быстрее приводит к формированию дыхательной недостаточности и инвалидизации больного. В связи с этим при верификации ЛАМ принципиально важно своевременно выявить имеющийся у пациента ТС, поскольку данный факт имеет большое прогностическое значение и определяет тактику ведения пациента.
ТС, или болезнь Бурневилля–Прингла, – системная наследственная дисплазия, обусловленная нарушением закладки эктодермального зародышевого листка, которая характеризуется комбинированным опухолевидным поражением кожи, головного мозга, глазных яблок, сердца, почек и легких [14]. ТС – аутосомно-доминантное заболевание c неполной пенетрантностью, обусловленное различными мутациями генов TSC1 и/или TSC2 (Tuberous Sclerosis Complex). В соответствии с существующей на сегодняшний день классификацией наследственных дисплазий ТС относится к факоматозам (phakos – от греч. чечевица, родимое пятно) – нейроэктодермальным заболеваниям, включающим кроме ТС такие редкие нозологические формы, как нейрофиброматоз, синдром Стерджа–Вебера и болезнь Гиппеля–Линдау.
ТС верифицируется в соответствие с критериями диагностики этого заболевания, которые подразделяются на «большие» и «малые» [15].
Большие диагностические критерии ТС
К большим критериям диагноза ТС относятся: ангиофиброматоз лица (щеки, спинка носа); подногтевые фибромы; три пятна гипопигментации и более, полиоз; участки в виде шагреневых бляшек; множественные гамартомные узелки на сетчатке; бугорки в коре больших полушарий; субэпендимальные узелки; субэпендимальная гигантоклеточная астроцитома; рабдомиома сердца; почечные ангиомиолипомы или ЛАМ.
Малые диагностические критерии ТС
Среди малых диагностических критериев ТС можно назвать множественные ямки на эмали зубов; гамартомные полипы прямой кишки; костные кисты; фиброматоз десен; непочечные гамартомы; ахроматические пятна на сетчатке; кожные проявления в виде конфетти (мелкие круглые пятна); множественные кисты почек; миграция белого вещества мозга в виде линий луча.
Диагноз ТС считается неоспоримым при наличии 2 больших критериев или 1 большого и 2 малых. Вероятный диагноз ставится при наличии 1 большого и 1 малого критерия. Диагноз ТС считается сомнительным при наличии 1 большого критерия.
В отличие от ЛАМ, которым страдают преимущественно женщины, ТС встречается с одинаковой частотой у лиц обоих полов. Распространенность ТС значительно выше, чем распространенность ЛАМ, и в случае диагностики в детском возрасте варьирует от 1 на 6800 до 1 на 17 300 [15]. Примерно 1/3 взрослых пациентов с ТС имеют рентгенологические признаки ЛАМ [16, 17]. Столь частая распространенность ЛАМ среди лиц, страдающих ТС, естественно, наводит на мысль об этиологическом и/или патогенетическом родстве этих заболеваний.
Этиология ЛАМ пока до конца не изучена, однако установлено, что пациенты, страдающие ЛАМ, имеют мутации в тех же генах, что и лица с ТС, – в генах TSC1 и TSC2 [18]. Следует отметить, что мутации в генах ТС обнаруживаются не только при ЛАМ, ассоциированном с ТС, но и при спорадическом его варианте, т.е. при отсутствии болезни Бурневилля–Прингла. Наличие генных мутаций, безусловно, свидетельствует в пользу генетической детерминированности ЛАМ. Однако в отличие от ТС, передающегося по наследству, передача ЛАМ от матери к дочери не описана [11]. Все эти факты позволяют сделать вывод о том, что обе эти нозологические формы являются генетическими заболеваниями, поскольку обусловлены мутациями в одних и тех же генах ТС (TSC1 и TSC2), но при ТС мутации в этих генах являются гаметическими, т.е. возникают в гаметах и, следовательно, передаются по наследству, а при ЛАМ мутации в тех же генах являются соматическими (т.е. возникают не в гаметах, а в клетках тканей организма), вследствие чего и не передаются последующим поколениям. Таким образом, хотя ЛАМ, как и ТС, является генетической нозологией, в отличие от последнего он не является наследственным заболеванием.
Ген TSC1 кодирует синтез белка гамартина, а ген TSC2 – белка туберина. Гамартин и туберин образуют гамартин-тубериновый комплекс, который является ключевым посредником в суммировании информации о ростовой стимуляции и подавляет чрезмерный клеточный рост через mTOR (мишени для иммуносупрессанта рапамицина у млекопитающих) [15]. При ТС возникают инактивирующие мутации в обоих генах-супрессорах – TSC1 и/или TSC2, что приводит к снижению активности гамартин-туберинового комплекса, угнетающего избыточный клеточный рост, а следовательно, препятствующего росту гамартом. Для ЛАМ более характерны мутации в гене TSC2 [19]. Наличие дефекта гена TSC2 предположительно является причиной аномального ответа (роста) ЛАМ-клеток (атипичных гладкомышечных и эпителиоидных) на женские половые гормоны [15]. Эстрогены регулируют транскрипцию многих генов, кроме этого, они могут играть роль стимула в пролиферации и миграции гладкомышечных клеток в другие органы и ткани. У больных ЛАМ выявляются как точечные мутации гена TSC2, так и мутации, связанные с утратой гетерозиготности локуса TSC2, приводящие к потере функции подавления опухолевого роста и, следовательно, бесконтрольной пролиферации клеток («двойной удар по генам-супрессорам опухолей»).
Несмотря на выявленные мутации в генах TSC1 и TSC2, патогенез ЛАМ до конца не известен. Суммируя накопленные на сегодняшний день данные об этом заболевании, с патогенетической точки зрения ЛАМ можно рассматривать как многофокусный гамартомный (опухолеподобный) процесс, обусловленный генетическим дефектом, который определяет аномальный ответ мышечных клеток на женские половые гормоны, возможно, играющие важную роль в прогрессировании заболевания.
В действительности об опухолеподобном характере ЛАМ свидетельствуют некоторые характерные для злокачественных новообразований черты, присущие этому заболеванию. Классическими признаками злокачественности служат: 1) автономно пролиферирующий бессмертный клон клеток, непрерывно эволюционирующий в сторону независимости от контроля организма; 2) инвазия и 3) метастазирование [20].
1. Морфологический субстрат ЛАМ – это до некоторой степени бесконтрольный опухолевоподобный рост ЛАМ-клеток в интерстиции легких, лимфатических сосудах и узлах [21]. ЛАМ-клетки представляют собой неупорядоченно пролиферирующие клетки двух типов – эпителиоидные (внешне напоминающие эпителиальные) и гладкомышечные клетки. Видовая принадлежность этих клеток пока не установлена, поскольку, с одной стороны, они дают, как и полагается гладкомышечным клеткам, положительную реакцию на актин, виментин и десмин, а с другой – экспрессируют белок премеланоцита. Отсутствие фигур митоза исключает подозрение на злокачественный характер этой пролиферации [22].
2. В пользу «инвазивности» роста ЛАМ-клеток свидетельствует повышенное содержание в них SRF (serum response factor). SRF – сывороточный ответный фактор, увеличивающий экспрессию матриксных металлопротеиназ (ММП) 2-го (коллагеназа IV типа) и 14-го (мембраносвязанная ММП) типов, содержание которых при ЛАМ повышается. Как известно, степень инвазивного роста и метастазирования опухолевых клеток определяется их способностью расщеплять компоненты экстрацеллюлярного матрикса, такие как базальные мембраны и компоненты межтканевой стромы (коллаген, эластин, ламинин и др.), что достигается с помощью повышенного содержания ММП [23]. В отличие от многих протеолитических ферментов, способных расщеплять отдельные компоненты экстрацеллюлярного матрикса, ММП разрушают все его структуры, что и позволяет опухолевым клеткам внедряться на территорию других тканей. Следует добавить, что наряду с увеличением экспрессии ММП при ЛАМ отмечается снижение уровня их тканевых ингибиторов 3-го типа (TIMP-3). Однако следует отметить, что повышение уровня ММП также характерно и для других интерстициальных заболеваний легких, таких как идиопатический легочный фиброз и саркоидоз [24].
3. Говоря о ЛАМ как о гамартомном (опухолеподобном) заболевании, способном к метастазированию, можно упомянуть о возникновении ЛАМ в донорском легком после трансплантации [25, 26]. Некоторые исследователи рассматривают поражение легких при ЛАМ как следствие метастазирования из других органов, в которых выявляются сходные изменения (почки, лимфатические узлы) [19, 27].
К фактам, свидетельствующим о важной роли эндокринных воздействий, в первую очередь женских половых гормонов, можно отнести возникновение ЛАМ у женщин, преимущественно детородного возраста; дебют и обострение ЛАМ могут быть спровоцированы лечением эстрогенами; во время менструации или беременности наблюдается клиническое ухудшение, проявляющееся в нарастании одышки и развитии пневмотораксов, нередко рецидивирующих; после наступления менопаузы или удаления яичников прогрессирование заболевания в ряде случаев замедляется. ЛАМ часто сочетается с фибро- или лейомиомой матки [28–30]. В 80% случаев на ЛАМ-клетках обнаруживаются поверхностные PRs- (прогестероновые) и ERs- (эстрогеновые) рецепторы, что подтверждает гормональную зависимость заболевания [7].
Резюмируя сказанное, можно говорить о ведущей роли популяции ЛАМ-клеток в патогенезе заболевания. ЛАМ-клетки обладают фенотипом, способным к бесконтрольному росту, экспрессии гормональных рецепторов и протеаз, а также склонным к метастатическому поведению. Пролиферация ЛАМ-клеток преимущественно имеет место в легких и в лимфатической системе, что и определяет клинические проявления этого заболевания, которые можно разделить на легочные и внелегочные соответственно.
В легких пролиферация ЛАМ-клеток выявляется вокруг бронхиол, артерий, вен, в лимфатических сосудах, а также в межальвеолярных перегородках и плевре [22, 31]. Сужение просвета мелких бронхиол, вероятно, за счет формирования «воздушных ловушек» приводит к гиперинфляции с последующим образованием мелких кист, что и объясняет макроскопическую картину микрокистозной перестройки паренхимы. При разрыве кист, расположенных подплеврально, развиваются пневмотораксы, часто рецидивирующие. В финале такой перестройки легочной паренхимы развивается дыхательная недостаточность, клинически проявляющаяся прогрессирующей одышкой. Разрастание ЛАМ-клеток в сосудах малого круга также играет значимую роль в патогенезе и клинической картине этого заболевания. Сужение просвета артериол приводит к значительному росту сосудистого сопротивления в малом круге кровообращения и, следовательно, к легочной гипертензии с последующим закономерным формированием легочного сердца. Разрастание ЛАМ-клеток вокруг венул приводит к их компрессии с сужением просвета и повышением давления внутри сосудов, к нарушению целостности сосудистой стенки, геморрагиям, гемосидерозу, а клинически проявляется кровохарканьем [21, 32]. При сдавлении лимфатических сосудов легких и плевры узелками пролиферирующих ЛАМ-клеток с их последующей обтурацией и разрывами развиваются хилоторакс и хилоптоэ. Хилоторакс имеет место у 28% больных ЛАМ [4], в то время как в общей структуре плевральных выпотов по частоте встречаемости он занимает одно из последних мест. При подобном поражении лимфатических сосудов брюшной полости, таза и ретроперитонеального пространства развивается хилезный асцит. Специфическое для ЛАМ поражение лимфатической системы – развитие лимфангиолейомиом – скопление увеличенных и кистозно-расширенных лимфатических сосудов, за счет инфильтрации ЛАМ-клетками. Поражение лимфатической системы при ЛАМ может проявляться лимфаденопатией. Кроме того, внелегочными проявлениями ЛАМ являются ангиомиолипомы почек и менингиомы.
При анализе симптомов, которые встречаются при ЛАМ, можно говорить о том, что в клинической картине этого заболевания доминирующей является прогрессирующая одышка с постепенным снижением толерантности к физической нагрузке, встречающаяся в 94% случаев [4] и являющаяся проявлением дыхательной недостаточности. Спонтанный пневмоторакс развивается в 80–81% случаев [4, 33] и может быть первым проявлением заболевания. У 41% больных были жалобы на кашель, в основном непродуктивного характера. Кровохарканье и боли в грудной клетке предъявляли 44 и 34% пациентов соответственно, а у 28% пациентов был обнаружен хилоторакс [4]. Кроме этого, описано развитие стридорозного дыхания и такие внелегочные поражения, как ангиомиолипомы почек. Возможно бессимптомное течение заболевания.
Диагноз ЛАМ устанавливают в соответствии с разработанными критериями и может быть оценен как достоверный, вероятный или сомнительный [12].
Диагностические критерии
лимфангиолейомиоматоза
Достоверный диагноз ЛАМ может быть поставлен, если выявляются:
1. «Характерные для ЛАМ легких» или «совместимые с ЛАМ легких» КТ-признаки и биопсия легких, соответствующая морфологическим критериям для ЛАМ
или
2. «Характерные для ЛАМ легких» КТ-признаки и любой из следующих:
• ангиомиолипома (почки);
• хилезный выпот в грудной или брюшной полости;
• лимфангиолейомиома или поражение лимфоузлов, характерное для ЛАМ;
• определенный или возможный ТС.
Вероятный диагноз ЛАМ верифицируется, если имеются следующие проявления:
1. «Характерные для ЛАМ легких» КТ-признаки и клиническая картина
или
2. «Совместимые с ЛАМ легких» КТ-признаки и любой из следующих: ангиомиолипома почки, хилезный выпот в грудной или брюшной полости.
Сомнительный диагноз ЛАМ базируется лишь на КТ-признаках:
1. «Характерные для ЛАМ легких» или «совместимые с ЛАМ легких» КТ-признаки.
По степени специфичности для ЛАМ рентгенологические критерии подразделены на «характерные для ЛАМ легких» и «совместимые с ЛАМ легких».
Характерные для ЛАМ легких КТ-признаки являются более специфичными для ЛАМ и включают множественные тонкостенные круглые хорошо дифференцируемые воздушные кисты при сохранном или увеличенном объеме легких без других значимых поражений, отсутствие признаков других интерстициальных болезней легких, за исключением маленьких узелков, которые наиболее часто служат КТ-проявлением мультифокальной микроузловой гиперплазии пневмоцитов у пациентов с ТС (как без, так и в сочетании с ЛАМ).
К совместимым с ЛАМ легких изменениям, выявляемым при КТВР, относят наличие только небольшого количества (от 2 до 10) таких кист, размером до 30 мм в диаметре, при соблюдении прочих перечисленных выше условий.
К наиболее информативным диагностическим методам при ЛАМ относят КТВР и морфологическое исследование ткани легкого, поскольку для этих методов разработаны наиболее достоверные критерии. Сочетание положительных результатов обоих этих исследований позволяют убедительно диагностировать ЛАМ [12].
По данным большинства имеющихся на сегодняшний день исследований, к «золотому стандарту» диагностики относят морфологическое исследование биоптата ткани легкого. Критериями диагноза, на основании наличия которых опытный патологоанатом может диагностировать ЛАМ, служат одновременное присутствие множественных мелких воздушных кист и наличие мультифокальной узловой пролиферации двух основных видов ЛАМ-клеток – незрелых гладкомышечных и периваскулярных эпителиоидных клеток. Поскольку опыт морфолога в диагностике столь редкого заболевания не всегда позволяет сделать однозначные выводы, а также в случаях, когда морфологическая картина, особенно на ранних стадиях заболевания, не вполне специфична, целесообразно проведение дополнительного иммуногистохимического исследования для выявления экспрессии α-гладкомышечного актина и НМВ-45. Кроме этого, полезным дополнением при постановке морфологического диагноза может быть определение рецепторов к эстрогену и прогестерону.
Тем не менее в соответствии с разработанными на сегодняшний день критериями диагностики ЛАМ достоверный диагноз может быть верифицирован и без проведения исследования биоптата [12, 13].
Распространение в последние годы в клинической практике КТВР позволило выявить рентгенологические маркеры поражения легких при ЛАМ, что сделало этот метод исследования обязательным в диагностике данного заболевания, поскольку на основании убедительных КТ-признаков в сочетании с соответствующей клинической картиной ЛАМ можно диагностировать с высокой достоверностью и без проведения биопсии легкого [12]. В тех случаях, когда достоверный диагноз ЛАМ ставится без результатов биопсии легких, помимо результатов КТВР большое значение придается как легочным, так и внелегочным клиническим проявлениям заболевания.
Как указывалось выше, внелегочными критериями диагностики ЛАМ являются наличие ангиомиолипомы почки (подтверждается при наличии КТ- и/или морфологических критериев опухоли), хилезного выпота в плевральной или брюшной полости (характер выпота устанавливается либо визуально, либо по биохимическим маркерам), лимфангиолейомиомы или лимфаденопатии, характерной для ЛАМ (с обязательной морфологической верификацией) или достоверного или вероятного диагноза ТС, который устанавливается в соответствии с критериями диагноза болезни Бурневилля–Прингла [15]. В соответствии с разработанными критериями диагностики вероятный диагноз ЛАМ может быть поставлен при сочетании характерных КТ-признаков с клинической картиной, включающей множественный и/или билатеральный пневмоторакс и/или соответствующие ЛАМ изменения функции легких, которые состоят в преимущественном снижении TLCO и в меньшей степени объема форсированного выдоха за 1-ю секунду при сохранных легочных объемах [12, 13].
На сегодняшний день эффективного лечения ЛАМ не существует. Пациентам главным образом проводится симптоматическая терапия: бронходилататоры, кислородотерапия. Предпринимаются попытки патогенетической терапии, основанные на данных о неблагоприятном влиянии эстрогенов на течение заболевания: антиэстрогенные препараты (тамоксифен, прогестерон) и редукция синтеза эстрогенов (оофорэктомия, радиоабляция яичников). Убедительных данных об эффективности этих методов лечения не получено, но наиболее целесообразным считается отмена эстрогенных препаратов, в том случае, если лечение ими проводится в связи с другими заболеваниями, а также предохранение от беременности. Вопрос о трансплантации легких, принимая во внимание возможность рецидива в трансплантате, также пока остается весьма дискутабельным. На сегодняшний день показаниями для трансплантации являются III или IV функциональный класс по NYHA с гипоксемией в покое, тяжелым нарушением легочной функции и дееспособности [12].
Лимфангиолейомиоматоз
Лимфангиомиоматоз.
Лимфангиолейомиоматоз
Продолжение.
Лимфангиомиоматоз. Лимфангиолейомиоматоз
ID: 19525 Lymphangioleiomyomatosis Dr Mark Holland - 19 Sep 2012 Coarse lung markings with small cysts throughout both lungs involving th...
ID: 24010 Lymphangioleiomyomatosis Dr Iqbal Naseem - 23 Jul 2013 LAM can associated with tuberous sclerosis complex or it can be isolate...
ID: 24127 Lymphangiomatosis Dr Jeremy Jones - 29 Jul 2013 Features here are lymphatic thickening and lymphangiomatosis which was c...
ID: 9464 Lymphangiomyomatosis Dr Andrew Dixon - 22 Apr 2010 There is diffuse cystic lung disease where the cysts are round, thin wal...
Случаи и цифры
Дифференциальная диагностика изображения