Туберозный склероз, или синдром Бурневиля - Прингля (теория).
М. Ю. Дорофеева, О. С. Страхова, О. В. Катышева, Э. К. Осипова, О. И. Чумак, М. В. Добрынина
Туберозный склероз - генетически детерминированное заболевание, относится к группе нейроэктодермальных нарушений, характеризуется поражением нервной системы, кожи и наличием доброкачественных опухолей (гамартом) в различных органах.
Первое описание клинического случая было сделано в 1862 г. F. von Recklinghausen. В 1880 г. D.-M. Bourneville подробно описал изменения, возникающие в головном мозге при этом заболевании.
Частота туберозного склероза составляет 1 : 30 000 населения. Распространенность среди новорожденных варьирует от 1 : 6000 до 1:10 000.
Туберозный склероз наследуется по аутосомно-доминантному типу. Большинство случаев заболевания (80%) является следствием мутации de novo. Болезнь отличается варьирующей экспрессивностью и почти 100%-ной пенетрантностью. Развитие туберозного склероза определяется двумя генами, локализованными в участке 34 длинного плеча 9-й хромосомы (туберозный склероз 1-го типа - TSC1, кодирует белок гамартин) и в участке 13 короткого плеча 16-й хромосомы (туберозный склероз 2-го типа - TSC2, кодирует белок туберин) [32].
В 1998 г. были приняты диагностические критерии заболевания. Несомненный диагноз туберозного склероза: два или один первичный признак + два вторичных признака. Возможный диагноз: один первичный признак + один вторичный признак. Предположительный диагноз: или один первичный признак, или два (и больше) вторичных признака.
Клиническая характеристика
Кожные изменения при туберозном склерозе представлены гипопигментными пятнами, ангиофибромами лица, участками «шагреневой кожи», околоногтевыми фибромами, фиброзными бляшками, белыми прядями волос.
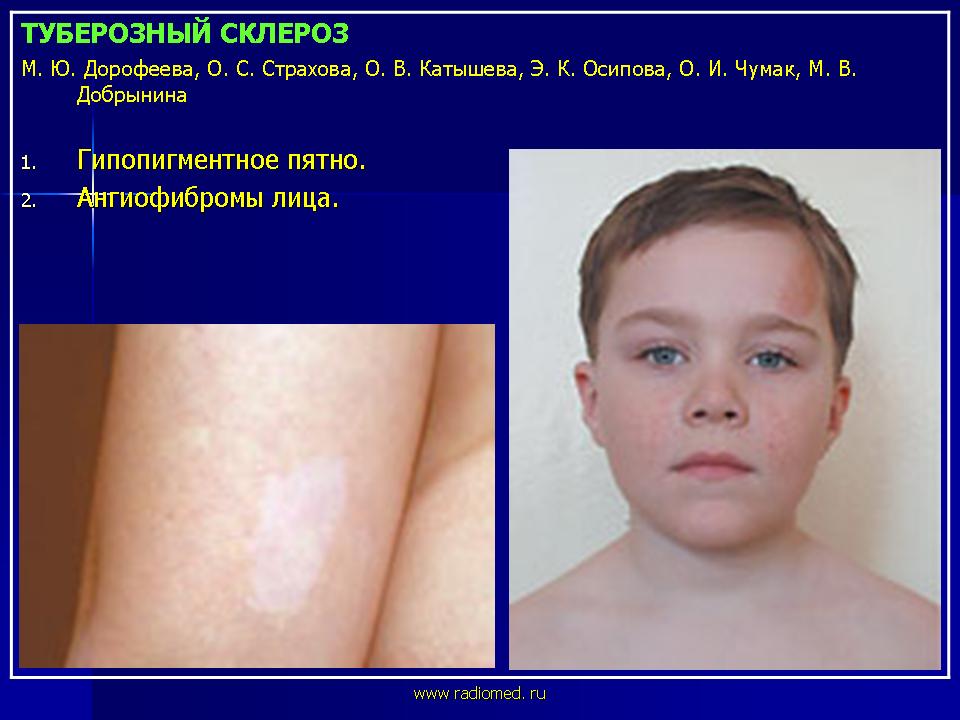
Гипопигментные пятна являются одним из наиболее частых кожных проявлений туберозного склероза. Они встречаются в 90% случаев и нередко обнаруживаются с рождения, являясь одним из манифестных признаков заболевания. С возрастом наблюдается тенденция к увеличению их числа. Гипопигментные пятна при туберозном склерозе локализуются преимущественно на туловище и ягодицах. Характерной их особенностью является асимметричность расположения. Отмечена вариабельность числа, размера и формы пятен.
С младенчества могут выявляться белые пряди волос, ресниц и бровей, которые, как и гипопигментные пятна, являются характерным признаком туберозного склероза.
Наряду с гипопигментными пятнами при туберозном склерозе в 15,4% случаев встречаются пигментные пятна цвета «кофе с молоком», что не превышает средних популяционных значений.
Ангиофибромы лица - облигатный признак туберозного склероза - наблюдаются в 47-90% случаев и появляются, как правило, после 4 лет. Внешне они представляют собой папулы или узлы розового или красного цвета с гладкой, блестящей поверхностью. Ангиофибромы располагаются на лице симметрично, с двух сторон - на щеках и на носу по типу «крыльев бабочки», а также на подбородке.
«Шагреневая кожа» (peau chagrin в переводе с франц. - «недубленая, грубая, жесткая кожа») - облигатный признак туберозного склероза, встречается в 21-68% случаев. В большинстве случаев «шагреневая кожа» появляется на втором десятилетии жизни. Участки «шагреневой кожи» наблюдаются преимущественно в пояснично-крестцовой области, имеют плотную консистенцию, желтовато-коричневый или розовый цвет, умеренно выступают над поверхностью окружающей кожи. Количество участков «шагреневой кожи» вариабельно, но чаще они бывают единичными. Размер их колеблется от нескольких миллиметров до 10 см и более.
Фиброзные бляшки встречаются у 25% больных с туберозным склерозом и также являются облигатным признаком заболевания. Фиброзные бляшки имеют бежевый цвет, шероховаты на ощупь и несколько выступают над окружающей кожей. Они часто появляются уже на первом году жизни и являются, таким образом, одним из первых клинических симптомов заболевания. Чаще всего фиброзные бляшки локализуются на лбу. Размеры и число бляшек могут варьировать.
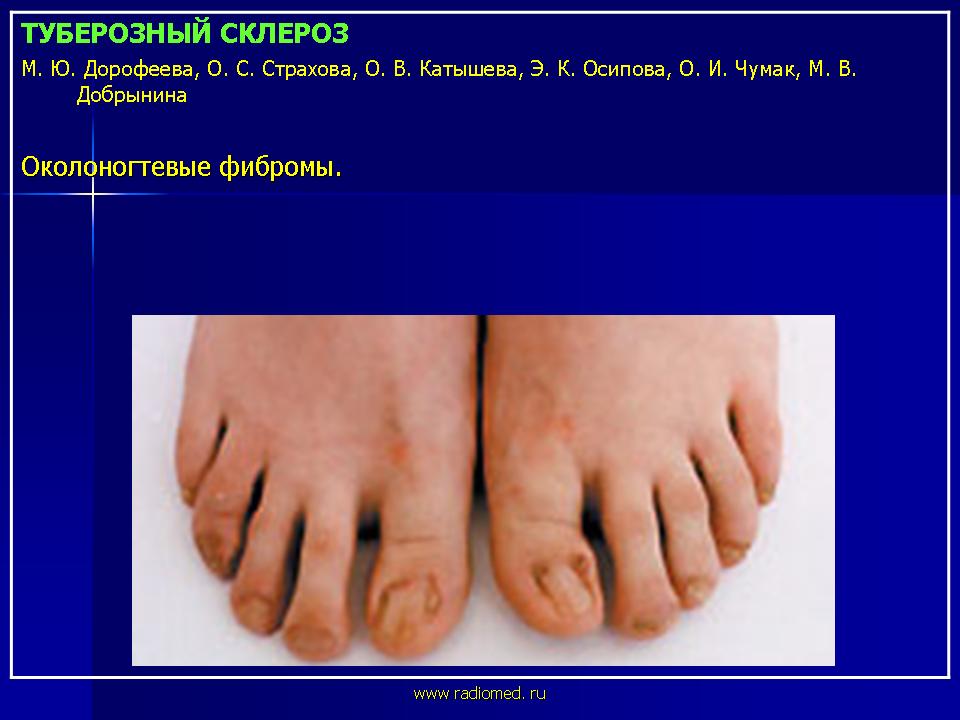
Околоногтевые фибромы - облигатный признак туберозного склероза (рис. 3). Они представляют собой тусклые, красные либо мясного цвета папулы или узлы, растущие от ногтевого ложа или вокруг ногтевой пластинки. Околоногтевые фибромы встречаются в 17-52% случаев. В большинстве случаев околоногтевые фибромы появляются на втором десятилетии жизни. Наиболее часто они локализуются на ногах. Размер их варьирует от 1 мм до 1 см в диаметре. Наличие околоногтевых фибром более характерно для женщин.
Мягкие фибромы встречаются у 30% больных. Они представляют собой множественные или единичные мягкие образования на ножках, мешотчатой формы, растущие на шее, туловище и конечностях (molluscum fibrosum pendulum). Другой вариант мягких фибром представляет собой множественные, несколько приподнятые над поверхностью кожи (и такого же цвета) мелкие образования, размером меньше булавочной головки, располагающиеся на туловище и шее и напоминающие гусиную кожу.
Наиболее типичными поражениями головного мозга при туберозном склерозе являются корковые туберы, субэпендимальные узлы и аномалии белого вещества мозга.
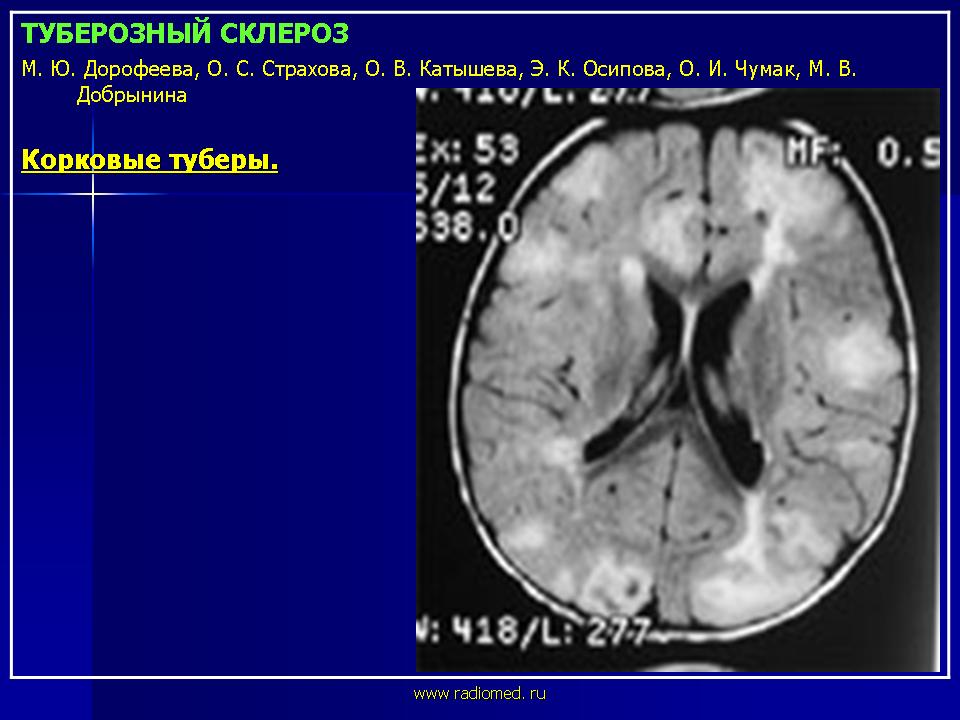
Корковые туберы различаются по своим размерам, локализации, консистенции и форме. Размер корковых туберов варьирует от нескольких миллиметров до нескольких сантиметров. Корковые туберы располагаются в виде выступов над единичной или прилегающими бороздами. Они расширяют борозду и сглаживают грань между серым и белым веществом. Туберы могут быть как единичными, так и множественными, имеют диффузную локализацию. Кальцификация туберов отмечается в 54% случаев и увеличивается с возрастом больных.
Своевременное выявление корковых туберов и кальцификатов мозга очень важно для диагностики туберозного склероза. Наибольшую значимость в верификации туберов при обследовании больных имеет магнитно-резонансная томография (МРТ), которая позволяет визуализировать туберы в 95% случаев.
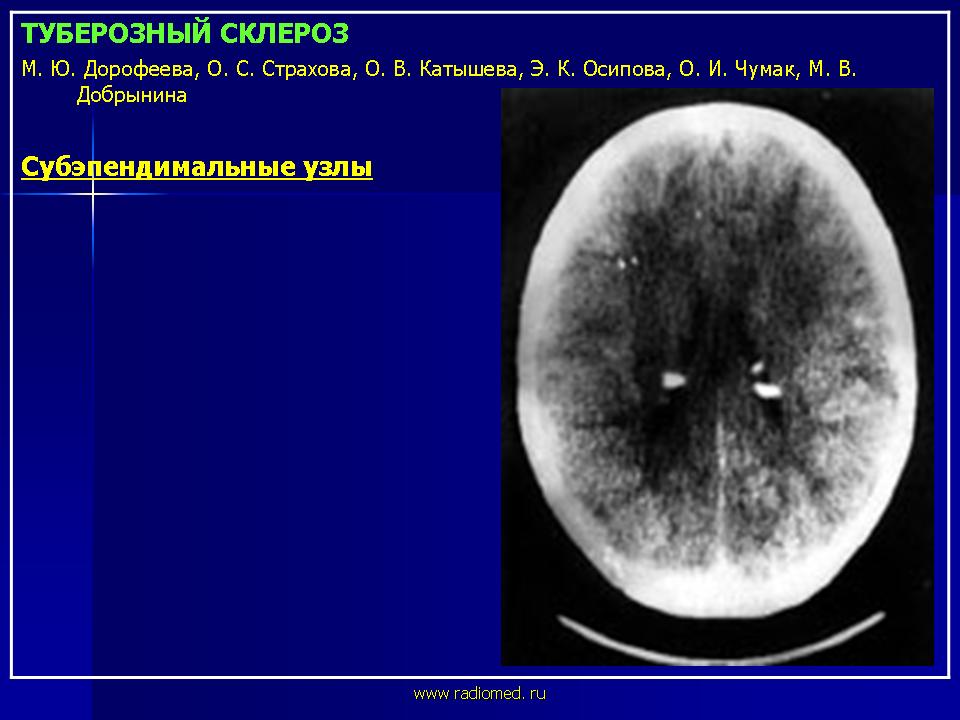
Субэпендимальные узлы встречаются в 95% случаев и выявляются как при компьютерном томографическом (КТ), так и при МРТ-исследованиях мозга. Субэпендимальные узлы в большинстве случаев множественные, прилегающие друг к другу. Локализуются, как правило, в стенках боковых желудочков, реже - в стенках III и IV желудочков мозга. При локализации в стенках боковых желудочков субэпендимальные узлы глубинной частью могут внедряться в хвостатое ядро или таламус. Форма субэпендимальных узлов обычно округлая или вытянутая. По мере роста ребенка в субэпендимальных узлах происходит постепенное отложение кальция. На компьютерных томограммах доминирующим признаком заболевания являются множественные, полностью или частично кальцифицированные, субэпендимальные узлы округлой формы, локализующиеся в стенках боковых желудочков. КТ-исследование более чувствительно, когда речь идет о выявлении кальцифицированных субэпендимальных узлов.
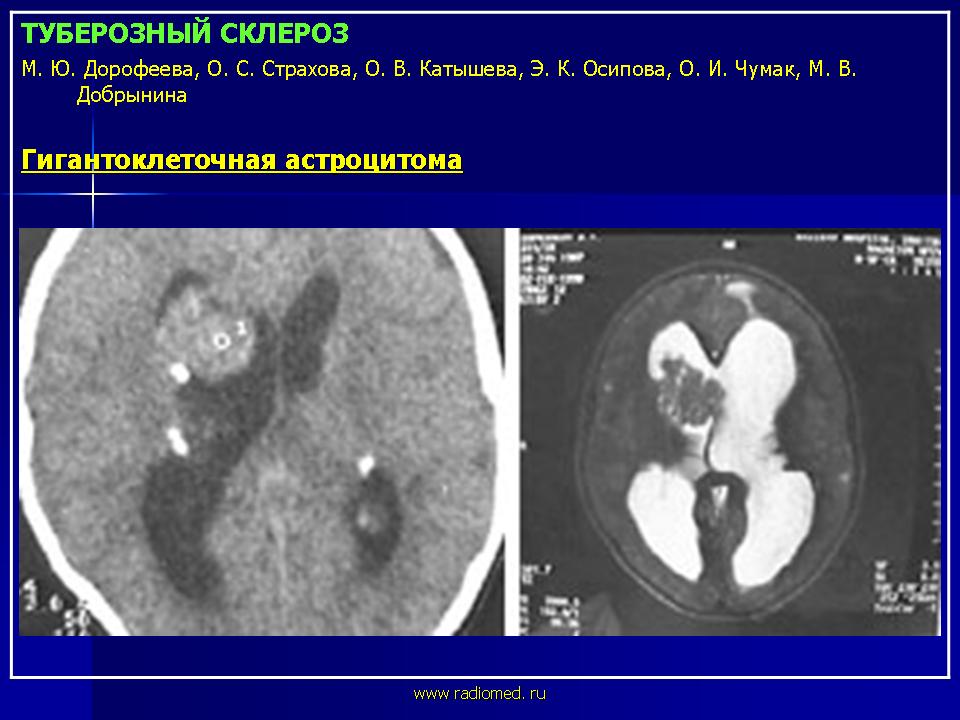
Субэпендимальные узлы нередко трансформируются в гигантоклеточную астроцитому и выявляются у 10-15% больных [47, 51]. Субэпендимальные гигантоклеточные астроцитомы манифестируют обычно между 5 и 10 годами жизни (средний возраст в момент выявления опухоли - 13 лет), как правило, имеют тенденцию к росту и всегда локализуются у межжелудочкового отверстия. Для диагностики гигантоклеточных астроцитом применяются как КТ-, так и МРТ-исследования. В клинической практике нередко используют оба нейрофизиологических метода.
Учитывая тот факт, что при туберозном склерозе гигантоклеточные астроцитомы наблюдаются относительно часто, рекомендуется динамическое проведение нейрорадиологических исследований у больных детей, не реже 1 раза в 2 года. В случае появления таких клинических симптомов, как головная боль, рвота, ухудшение зрения, необходимо экстренное проведение нейрорадиологических исследований.
Поражение белого вещества головного мозга при туберозном склерозе характеризуется появлением своеобразных островков, состоящих из групп гетеротопических кластерных клеток и располагающихся вдоль линий, соединяющих эпендиму стенок желудочков и туберы. Данные линии соответствуют нормальным миграционным путям спонгиобластов во время эмбриогенеза.
У 10% больных при туберозном склерозе описаны поражения мозжечка.
Поражения нервной системы являются доминирующими в клинической картине туберозного склероза. Наиболее характерны судорожные пароксизмы, умственная отсталость, нарушения поведения, изменения в цикле «сон-бодрствование».
Судорожные пароксизмы - один из наиболее значимых симптомов туберозного склероза - наблюдаются у 80-92% больных [19, 38] и чаще всего являются манифестным симптомом заболевания. Эпилептические пароксизмы при туберозном склерозе нередко резистентны к противосудорожной терапии, могут приводить к развитию нарушений интеллекта и поведения и являются одной из главных причин инвалидности у детей с туберозным склерозом. P. Curatolo [17] отмечено, что среди факторов, детерминирующих резистентность к противосудорожной терапии, наибольшее значение имеют: дебют в возрасте до 1 года, наличие нескольких типов приступов, высокая частота приступов, изменение характера приступов с течением заболевания.
Умственная отсталость при туберозном склерозе наблюдается в 48% случаев [10, 18, 28] и варьирует от умеренной до глубокой степени. Одной из основных причин, которые определяют ее возникновение, являются судороги, возникающие на первом году жизни. Нарушение интеллекта при туберозном склерозе сочетается с изменениями поведения в виде аутизма, гиперактивности, агрессивности.
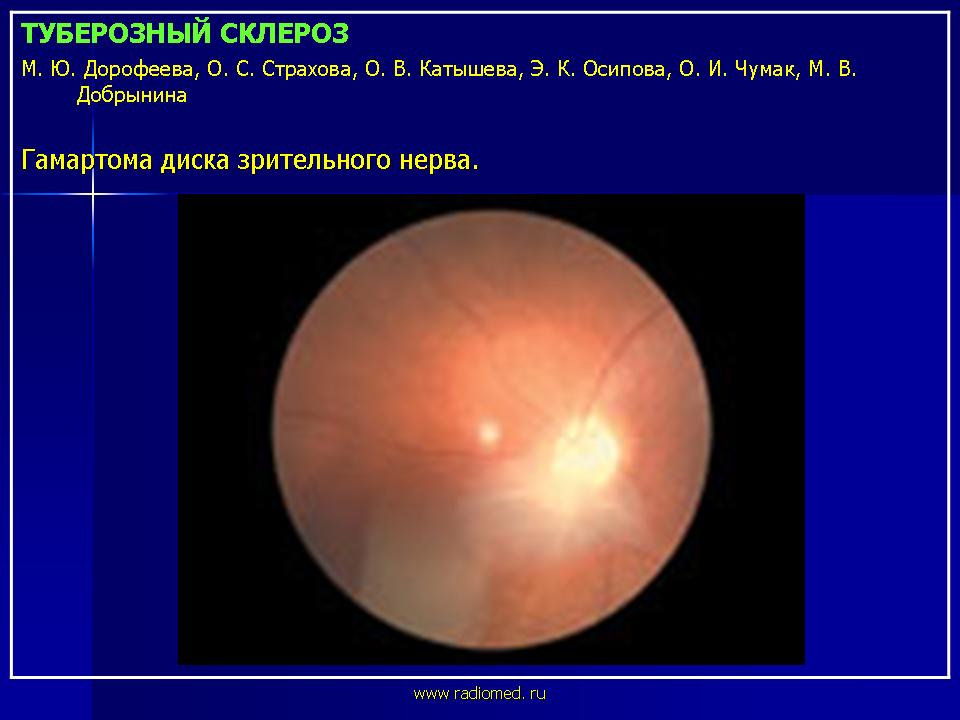
Поражение органов зрения при туберозном склерозе характеризуется появлением гамартом сетчатки и зрительного нерва, которые выявляются примерно у 50% больных. У половины больных они множественные. Выделяют три наиболее типичных варианта гамартом сетчатки. При первом, наиболее распространенном варианте гамартомы имеют нежную, относительно плоскую и гладкую поверхность, оранжево-розовый цвет, округлую или овальную форму, локализуются преимущественно в поверхностных слоях сетчатки. При втором - гамартомы имеют узловатый вид и напоминают тутовую ягоду. Они белого цвета, кальцифицированные, светонепроницаемые. При третьем варианте гамартомы сочетают в себе признаки первых двух. Они имеют округлую форму с узловатым и кальцифицированным центром и полупрозрачной, гладкой периферией оранжево-розового цвета.
Клинические проявления гамартом наблюдаются крайне редко. Основным симптомом является прогрессирующее снижение зрения.
Изменения сердечно-сосудистой системы при туберозном склерозе проявляются развитием рабдомиом, которые нередко служат первым клиническим признаком туберозного склероза наряду с гипопигментными пятнами. Рабдомиомы встречаются в 30-60 % случаев и выявляются чаще у лиц мужского пола (соотношение 2 : 1). Наиболее высокая частота рабдомиомы сердца при туберозном склерозе наблюдается у новорожденных (у 21 из 23 детей) и детей грудного возраста (у 11 из 33 детей).
Рабдомиоматозные образования могут быть в виде одного узла или множественными. Они, как правило, локализуются в желудочках и имеют смешанный интраэкстрамуральный рост.
В очень редких случаях рабдомиомы могут локализоваться в предсердиях, исходя из межпредсердной перегородки. Рабдомиомы различаются по своим форме и размерам, которые варьируют от нескольких миллиметров до нескольких сантиметров. Опухоли имеют неправильную форму и всегда четко отделены от окружающих тканей.
Ультразвуковое исследование позволяет выявить опухоль сердца еще во время внутриутробного развития плода, начиная с 21 нед гестации. Во всех случаях внутриутробной диагностики опухоли у новорожденного должен быть исключен туберозный склероз даже при отсутствии семейного анамнеза.
МРТ-исследование сердца более информативно при определении степени прорастания миокарда, поскольку позволяет определить «демаркационную» линию опухоли, отделяющую от рабочего миокарда.
Клинические симптомы рабдомиом у новорожденных различны. При опухолях, диагностированных внутриутробно, в 4 случаях из 11 наблюдалась внутриутробная смерть плода. Примерно у 50% новорожденных опухоль может выявиться случайно при проведении планового эхокардиографического обследования по поводу туберозного склероза. Обычно эти опухоли не нарушают гемодинамику и не имеют выраженного интрамурального роста. Известны случаи диагностики рабдомиомы при обследовании новорожденных по поводу пароксизмальной тахикардии.
При массивных опухолях может наблюдаться внутриутробная смерть плода либо дети рождаются преждевременно с низкой оценкой по шкале Апгар, имеют распространенные отеки и выраженный цианоз. Имеются сообщения о случаях смерти новорожденных от застойной сердечной недостаточности.
Замечено, что рабдомиомы сердца, как правило, быстро увеличиваются во время второй половины беременности, в основном достигают максимальных величин к моменту рождения, а затем постепенно уменьшаются в размерах. Большинство рабдомиом исчезают бесследно. Спонтанная регрессия рабдомиом наблюдалась у детей младше 6 лет. После 6 лет опухоли обычно не исчезают, однако могут несколько уменьшаться в размере. Регресс опухолей может наблюдаться как в размере, так и в числе.

Вариант ультразвуковой картины почек и печени у ребенка с туберозным склерозом.
О.П. Калмыкова, С.Г. Бондаренко, Г.Г. Гальчук
Клиническая больница N 7, Волгоград, Россия
Введение
Туберозный склероз, или синдром Бурневиля-Прингля, впервые описанный в 1862 году, представляет собой генетическое заболевание, наследуемое по аутосомно-доминантному типу с двумя вариантами поражения генов. Первый вариант туберозного склероза развивается при мутации гена, расположенного на хромосоме 9q34, второй - в связи с мутацией другого гена на хромосоме 16q13. Частота заболевания в популяции - 1:10000.
В настоящее время известно, что мутация перечисленных генов является основой для развития гиперпластических процессов в связи со снижением синтеза белка туберина.
Основными клиническими симптомами являются полиморфные судорожные приступы, психические расстройства в виде снижения интеллекта вплоть до идиотии, узелковые высыпания на лице в виде бабочки (аденома Прингля), локальное поседение, кожные фибромы, гемангиоэктазии, участки по типу шагреневой кожи, доброкачественные опухоли в сердце (рабдомиомы), адипозогенитальный синдром, деформации грудной клетки, позвоночника и конечностей.
У взрослых пациентов при туберозном склерозе наиболее часто диагностируются ренальные ангиомиолипомы. Так Steiner и соавт. у 17 % с ангиомиолипомами почек обнаружили туберозный склероз, причем нередко отмечалось двустороннее поражение. В этом возрастном контингенте к другой по частоте патологии почек при туберозном склерозе относятся гамартомы, которые могут сочетаться с саркомой. В детском возрасте патогномоничным для туберозного склероза являются кистозные поражения почек, и, как правило, поражение носит двусторонний характер.
Скрининговые исследования печени у детей с туберозным склерозом, проведенные Jozwiak и соавт., обнаружили гамартому у 23,5 % пациентов детского возраста.
В представленном наблюдении приводим нетипичное поражение почек и печени у ребенка с туберозным склерозом.
Описание случая
Девочка, 9 лет, поступила в хирургическое отделение с жалобами на острые боли в животе. Из анамнеза было выяснено, что пациентка состоит на диспансерном учете в течение шести лет у невропатолога по поводу туберозного склероза. Страдала эпилептиформными припадками, последние три года отмечалась ремиссия. Неоднократно проходила обследование головного мозга: при ЯМР - томографии в затылочных, теменных и лобных долях определялись зоны диаметром до 10 мм., с нечеткими неровными контурами, распространяющимися как на белое, так и на серое вещество (гамартомы?) и субэпиндимальные узлы в боковых желудочках.
При поступлении состояние удовлетворительное, девочка правильного телосложения, повышенного питания. На лице ярко-красные плотные бугорки в виде бабочки (характерно, что на лице у матери пациентки аналогичные аденоматозные высыпания), кожа на туловище и конечностях покрыта обильными уртикарными элементами, на большом пальце левой кисти - изменения кожи по типу "шагреневой". Область головки лучевой кости деформирована, здесь же определяется опухолевидное образование плотноэластической консистенции. При доплерографии сосудов левого предплечья, выполненной год назад, были выявлены признаки дисциркуляции и сосудистой мальформации.
При эхокардиографии обнаружена ложная хорда левого желудочка.
Следует отметить, что при ультразвуковом исследовании печени и почек, выполненных год назад, каких-либо структурных изменений не определялось.
Заключение
В доступной нам литературе мы не встретили описания сочетанного поражения печени и почек, подобного представленному случаю. Если кистозная трансформация и гамартома относятся к наследственному дисэмбриогенезу, то приведенное клиническое наблюдение (с учетом характерного поражения головного мозга, кожи, печени и почек, динамики появления изменений) можно интерпретировать как диссиминированный фиброматоз.
Туберозный склероз.
Сакович Р. А. 1, Чиж Г. В. 2
1Республиканская клиническая психиатрическая больница, 2БелМАПО.
Туберозный склероз (ТС) - полисистемное генетическое заболевание, которое проявляется бугорковоподобными разрастаниями в веществе головного мозга, а также в других жизненно важных органах: коже, почках, глазах, легких, сердце, костях. Заболевание врожденное, длится на протяжении всей жизни. Частота распространения 1 на 7-10 тысяч человек.
ТС был впервые описан Recklinghausen в 1862 году. В 1880 году Bourneville детализировал выявляемые патологические данные и впервые применил термин туберозный склероз. Bourneville описал данную нозологию как «туберозный склероз церебральных волокон». Данные патологические изменения он выявил у 15 летней девочки, страдавшей эпилепсией, имевшей кожные проявления, а также снижение интеллекта.
ТС наследуется по аутосомно-доминантному типу с различной пенентрантностью; как спонтанная мутация встречается в 60-70% случаев (ни один из родителей не имеет ТС); при наличии ТС у одного из родителей - вероятность ТС у ребенка составляет 50 %. Генетические исследования выявили два гена ТС:
- TSC1 - хромосома 9 - открыт в 1987 году,
- TSC2 - хромосома 16 - открыт в 1992 году.
Исследователи полагают, что протеин туберин, который в норме подавляет опухолевый рост в организме, недостаточно вырабатывается у больных ТС.
Клинические проявления.
В зависимости от степени вовлечения в патологический процесс различных органов и систем тяжесть течения заболевания может широко варьировать. Типичными симптомами ТС является триада:
1. Эпилепсия. Частота встречаемости эпилептических припадков у пациентов с ТС колеблется от 70 до 90% (по данным различных источников). Могут иметь место различные типы эпилептических припадков: · Инфантильные спазмы, тонически клонические, тонические, акинетические, миоклонические, атипичные; в некоторых случаях возможно формирование эпилептического статуса. Отмечается, что эпилептические припадки наблюдаются у всех больных с нарушением интеллекта, и у 70% пациентов без нарушений.
2. Интеллектуальное снижение. Большинство пациентов имеют проблемы с развитием интеллекта. Умственная отсталость зависит от степени поражения корковых центров, наличия сопутствующих аномалий развития головного мозга, а также от тяжести течения эпиприпадков. У пациентов с ТС наблюдаются проблемы с поведением: некоторые дети гиперактивны, агрессивны, тревожны, в некоторых случаях страдают аутизмом.
3. Кожные знаки. Одним из первых проявлений являются гипопигментированные пятна на туловище и конечностях, выявляемые даже у новорожденных (могут быть ложно диагностированы как родимые пятна); встречаются в 83% случаев, лучше визуализируется в ультрафиолетовых лучах. Позднее характерно появление ангиофибромы на лице - около носа, на подбородке, щеках. Встречается в 50-80 % случаев; проявляется в виде сыпи, состоящей из пятен размером 0.1-1 см. Распространяясь по лицу могут принимать фигуру «бабочки». С течением времени пятна имеют тенденцию к слиянию и изменению цвета (коричневый, темно-красный). В старшем возрасте диагностируются пятна Shagreen (в 30-70% случаев), подногтевые фибромы и гипопигментации.
Следует отметить, что больной ТС вследствие разнообразия клинических проявлений и степени их выраженности может наблюдаться у различных специалистов. В данной ситуации важной задачей является выявление полисистемности поражения и детекция классической триады.
Морфологические изменения.
Разнообразие клинических проявлений ТС является результатом мальформации фиброзной и соединительной ткани в пораженных органах. Аномалии миграции играют важную роль в неврологических расстройствах. При микроскопическом исследовании бугорковоподобные разрастания включают избыточное количество клеток одного или нескольких типов.
Бугорки могут поражать одну или более извилин головного мозга, располагаться субэпендимально, реже - в стволе мозга, базальных ядрах, в зубчатых ядрах мозжечка; имеют размер 0.2-2 см. В некоторых случаях, в зависимости от локализации, размеров могут вызывать окклюзионную гидроцефалию (в 7-10 % случаев). С течением времени бугорки кальцифицируются; иногда бугорки могут подвергаться злокачественному перерождению в гигантоклеточную астроцитому, реже глиобластому.
Методы лучевой диагностики ТС. Интракраниальные поражения.
Первоначально ТС диагностировался с помощью краниографии (при выявлении интракраниальных интрацеребральных кальцинатов) и пневмоэнцефалографии (при выявлении деформации желудочковой системы, а также выявлении окклюзионной гидроцефалии).
С внедрением в практику рентгеновской компьютерной томографии (РКТ) выявление интракраниальных проявлений ТС и верификация клинических данных достигла 75-85%. Бугорки ТС на РК-томограммах выглядят в виде очагов округлой формы, размером 0.2-2 см с четкими наружными контурами. Имеют при кальцинации высокую плотность, локализуются интрацеребрально: в извилинах, кортикально, субкортикально, субэпендимально, реже - в стволе мозга, базальных ядрах, в зубчатых ядрах мозжечка; в некоторых случаях выявляется утолщение костей свода черепа, гипоплазия мозолистого тела. При проведении контрастирования очаги ТС, как правило, не накапливают контрастный препарат; накопление же свидетельствует о злокачественном перерождении очага. Диагностика некальцифицированных очагов с использованием РКТ затруднена. При выявлении признаков окклюзионной гидроцефалии показано введение контрастного препарата для выявления изоденсивных и злокачественно перерожденных очагов.
Магнитно-резонансная томография (МРТ) открыла новый виток в качественной и количественной диагностике ТС. При проведении краниальной МРТ выявляются различные патологические изменения:
- Интрацеребральные бугорки,
- Кортикальные гетеротопии,
- Дефекты демиелинизации,
- Окклюзионная гидроцефалия,
- Наличие сопутствующих аномалий развития.
При проведении МРТ больному ТС следует обращать внимание на:
1. Наличие субэпендимальных бугорков - наиболее специфичный показатель. Применяются импульсные последовательности взвешенных по Т1 (=Т1W=короткие TR/TE) и по Т2 (=Т2W=длинные TR/TE). На МР-томограммах взвешенных по Т1 определение очагов затруднено из-за их изоинтенсивности к серому веществу. Следует отметить, что кальцинированные очаги могут быть изоинтенсивны на МРТ, в особенности при использовании низкопольных МР-систем.
2. Кортикальные и субкортикальные очаги повышенной интенсивности сигнала в Т2W и изогипоинтенсивные в Т1W. Отмечено, что именно очаги указанной локализации в наибольшей степени влияют на наличие эпиприступов, снижения интеллекта. В визуализации данных очагов МРТ имеет преимущество перед РКТ. Отмечено, что кортикальные и субкортикальные очаги имеют тенденцию к более высокой интенсивности сигнала на Т2W в сравнении с субэпендимальными. Данный факт объясняется повышенным содержанием жидкости в кортикальных и субкортикальных очагах, а также повышенной кальцинацией субэпендимальных очагов.
3. Расширение и деформацию желудочковой системы (возможна верификация её причины и уровня), а также повышение интенсивности сигнала в Т2W и снижение в Т1W перивентрикулярно от белого вещества.
4. Выявление очагов демиелинизации, которые бывают гиперинтенсивными в Т2W и изоинтенсивными в Т1W. Выявляются не всегда. Предпочтение следует отдавать импульсным последовательностям с длинными TR/TE.
5. Нарушение нормальной корковой архитектоники.
6. Наличие сопутствующих аномалий развития, к примеру, интракраниальных аневризм. Образование аневризм сосудов связано с нарушением развития стенки артериальных сосудов.
Экстракраниальные изменения.
Как указывалось выше при ТС определяется вовлечение в процесс различных органов и систем организма: почек, сердца, печени, легких, 12-перстной кишки и др.
- Поражения почек встречаются в 60-80% случаев; к ним можно отнести ангиомиолипомы, единичные или множественные гамартомы, аневризмы почечных сосудов. Ангиомиолипома почек частая находка при ТС. Приведенная ниже Таблица помогает в проведении дифференциального анализа.
Таблица. Особенности ангиомиолипом при ТС и без него.
Ангиомиолипомы, ассоциированные с ТС
Ангиомиолипомы, не ассоциированные с ТС
двухсторонние
односторонние
множественные
единичные
встречаются в детском возрасте без половой акцентуации
встречаются у женщин среднего возраста
- Изменения в скелете отмечаются в 40-65% случаев. Проявлениями ТС является остеопороз, кистоподобные дефекты костей запястья, плюсны, фаланг, локальные периостальные утолщения тел трубчатых костей, участки склероза в позвонках, костях таза.
- Поражение сердца встречается не часто, однако в 45% случаев у пациентов с миокардиальной рабдомиомой выявляется ТС
- В 1% случаев определяется вовлечение в процесс легких в виде неспецифического ретикулярного инфильтрата.
Клинические примеры.
Больной К. 9 лет. Жалобы на наличие миоклонических судорожных припадков, задержку психоречевого развития. Кожа бледно-розовая, на лице, руках - туберозные бугорки. Печень +2 см, селезенка не пальпируется; в правой половине живота определяется образование. По данным РКТ головного мозга обнаружены субэпендимальные кальцинаты с четкими контурами и признаки внутренней гидроцефалии (Рис. 1). При проведении РКТ и УЗИ почек визуализируются двусторонние солидные опухоли размером до 82х90х126мм. (ангиомиолипома - по данным гистологического исследования). (Данные исследования предоставлены Республиканским научно-исследовательским цетром детской онкологии и гематологии; РК-томограф Tomoscan SR (Philips).
Больной У. 4 года. При поступлении жалобы на судорожные приступы, умственную отсталость, глубокую задержку психоречевого развития. Судороги кивательные, 1-2 серии в день. Ребенок ходит; СПР D<S, высокие. На теле выявлены депигментированные пятна размером до 12 мм. При проведении краниальной МРТ определяются очаги гиперинтенсивные в Т2W и изогипоинтенсивные в Т1W, расположенные преимущественно кортикально и субкортикально. Субэпендимальные очаги явно не визуализируются, возможно, вследствие кальцинации (Рис. 2). (Собственное наблюдение, исследование проведено на базе республиканской клинической психиатрической больницы; МР-томограф «Образ-2М» 0.14 Тл.).
Заключение.
Исходя из вышесказанного, можно рекомендовать РКТ и МРТ как методы выбора в диагностике ТС. РКТ эффективна в выявлении кальцифицированных интрацеребральных очагов у больных ТС. В отличие от РКТ, МРТ особенно эффективна в диагностировании некальцифицированных интракраниальных и экстракраниальных очагов поражения, а также в выявлении широкого спектра сопутствующей патологии при ТС.
Туберозный склероз: дефиниция, особенности клинического течения
Шнайдер Н.А., д.м.н., профессор, ГОУ ВПО «Красноярский государственный медицинский университет имени профессора В.Ф. Войно-Ясенецкого Министерства здравоохранения и социального развития», кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования
Международный неврологический журнал 2 (32) 2010 / Лекция /lecture/
Введение
Туберозный склероз (ТС) представляет собой форму моногенной наследственной патологии и встречается в популяции с частотой 1 : 50 000 населения. Он относится к условно выделенной группе заболеваний — так называемым нейрокожным (нейрокутанным) синдромам, к числу которых относятся факоматозы (греч. phakos — пятно) — экзодермальные дисплазии, объединенные генетически детерминированным нарушением роста и дифференцировки клеток зародышевых листков, а также общими звеньями патогенеза. К факоматозам преимущественно эктодермального происхождения относят ТС, а также нейрофиброматоз (болезнь Реклингхаузена), нейрокожный меланоз, синдром базальноклеточного невуса и другие наследственные заболевания.
Общие характерные признаки факоматозов:
— дебют в детском и молодом возрасте;
— наследственность отягощена (аутосомно-доминантный тип наследования встречается чаще, чем другие типы наследования);
— полисистемность поражения;
— высокий риск развития множественных опухолей головного мозга, спинного мозга и внутренних органов;
— многие факоматозы протекают со снижением интеллекта;
— повышен уровень стигматизации (стигмы дизэмбриогенеза, дизрафический статус);
— снижена средняя продолжительность жизни.
Трудности диагностики факоматозов связаны с выраженным клиническим полиморфизмом и возрастзависимым дебютом симптомов. Пациенты с факоматозами в течение жизни наблюдаются врачами практически всех специальностей, поэтому только информированность специалистов о характере и особенностях течения данных заболеваний, а также согласованность их диагностических и лечебных мероприятий могут обеспечить выбор правильной тактики ведения больных.
Среди факоматозов одним из наиболее тяжелых по течению и прогнозу является ТС (болезнь Бурневилля — Прингла), характеризующийся системным поражением внутренних органов, костей, глаз, кожи, нервной системы (за счет нарушения пролиферации, миграции и дифференциации клеток нейроглии). Все изменения при ТС имеют единую патоморфологическую основу — гамартоматозную пролиферацию. Изменения в головном мозге наиболее наглядно демонстрируют дизонтогенетическую теорию развития этого заболевания. Считается, что уже на ранних этапах развития (13–17 нед. внутриутробного развития) образуются аномальные гигантские нейроглиальные клетки, которые в процессе своей миграции могут останавливаться в нетипичных местах: субэпендимально, образуя субэпендимальные узлы; в белом веществе полушарий — нейрональные гетеротопии; субкортикально и в проекции коры — субкортикальные и кортикальные туберсы. Гигантские нейроглиальные клетки являются продуктом дисгенезии на ранних этапах развития; первичные же нарушения лежат в клетках герментативного матрикса, функция которых и контролируется генами. Все пациенты с ТС любого типа, а также их родственники, особенно первой степени родства, подлежат мультидисциплинарному динамическому наблюдению и обследованию в течение всей жизни. Только такой подход может обеспечить адекватную терапию и позволит проводить медико-генетическое консультирование в семьях, где выявлены пациенты с этим тяжелым мультисистемным заболеванием.
Краткая история
Заболевание было впервые описано Frederich von Recklinghausen в 1862 г. В 1880 г. французский врач D.-M. Bourneville детализировал выявляемые патологические изменения, развивающиеся в головном мозге, и впервые применил термин «туберозный склероз церебральных волокон». Данные патологические изменения он выявил у 15-летней девочки, страдавшей эпилепсией, имевшей кожные проявления, а также снижение интеллекта. В 1890 г. дерматолог John James Pringle впервые описал ангиофибромы лица у больных ТС, применив термин «конгенитальная аденома сальных желез» ( adenoma sebaceum , лат.), который в современной литературе известен как аденома Прингла. В 1908 г. Vogt описал классическую клиническую триаду ТС (так называемый комплекс туберозного склероза, или туберозно-склерозный комплекс ( tuberous sclerosis complex — TSC, англ.)): судорожные приступы, аденомы сальных желез и задержка умственного развития, которые в течение длительного времени считались патогномоничными для данного заболевания. Заболевание характеризовалось высокой ранней детской смертностью и снижением продолжительности жизни у взрослых людей. В 1921 г. офтальмолог J. van der Hoeve описал характерные для ТС изменения на глазном дне: симптом тутовой ягоды — факому.
Исследования, проводимые в последние годы, значительно расширили представления о диагностических критериях ТС. В настоящее время показано, что классическая триада (аденома сальных желез, умственная отсталость и эпилепсия) встречается только у 25 % пациентов. При легких формах ТС правильный диагноз обычно не устанавливается.
Дефиниция
Туберозный склероз (син.: эпилойя, болезнь Бурневилля — Прингла, центральный нейрономатоз, нейрокожный синдром типа Бурневилля, синдром себорейной аденомы, судорог и умственной отсталости ) — наследственное заболевание, патоморфологическим субстратом которого является гамартоматозная пролиферация различной локализации. Тип наследования — аутосомно-доминантный с неполной пенетрантностью (заболевание клинически проявляется в потомстве далеко не во всех случаях). Патологические признаки обладают также неполной варьирующей экспрессивностью: симптомы заболевания могут проявляться в различной степени или полностью отсутствовать. Тяжелые формы ТС чаще бывают спорадическими, тогда как легким формам заболевания более свойственно семейное проявление. Число случаев с отрицательным семейным анамнезом велико — 50–70 % (спорадические случаи ТС). Наличие четко очерченных опухолевых узлов в нескольких органах позволяет классифицировать ТС как множественный бластоз. Тенденция к появлению клеточной атипии специфична для бластозного процесса. Однако сам факт развития первичных опухолей в разных органах и одновременно из нескольких клеточных зачатков может указывать на генетическую неполноценность предсуществующих опухолевых структур. Подтверждением дизэмбриогенеза при ТС являются признаки недоразвития в участках коры головного мозга в виде дистопии клеточных слоев и аномальных нервных клеток.
Эпидемиология
Частота встречаемости ТС у детей и подростков составляет 1 случай на 6800–12 000 детского населения (A. Hunt, С. Shepherd, 1993), в том числе: 1 на 15 000 для детей до 5 лет, 1 случай на 6000 новорожденных. Распространенность заболевания для всех возрастов около 1 на 30 000–50 000. ТС чаще встречается, чем диагностируется, так как индивидуумы, имеющие факультативные, неспецифические клинические признаки, в большинстве случаев не учитываются, а больные с облигатными признаками и синдромами, не являющимися нозологическими формами, довольно часто курируются врачами разных специальностей.
Этиопатогенез
ТС — аутосомно-доминантное генетически гетерогенное заболевание с неполной пенетрантностью, вариабельной экспрессивностью и высокой частотой возникновения новых (спонтанных) мутаций, которые обнаруживаются в 68 % всех случаев, дебютирующих в раннем возрасте (P. Fleury et al., 1980; A. Hunt, R. Lindenbaum, 1984; J. Ahlsen, 1994). С 1987 по 1993 г. были обнаружены генные участки, локализованные на хромосомах 9 и 16, ответственные за ТС. В 1997 г. был обнаружен генный маркер заболевания на хромосоме 9 при TSC1, что в будущем может иметь важное значение для пренатальной диагностики заболевания. Причиной развития ТС являются инактивирующие мутации в одном из генов. В зависимости от дефектного гена выделяют 3 типа заболевания: TSC1 — 9q34 (*191100, ген гамартина TSC1, 50 % семейного туберозного склероза), TSC2 — 16р13.3 (191092, ген TSC2), TSC3 — 1 Iq23. Вероятность дополнительного локуса, ответственного за развитие ТС, на хромосомах 11 и 12 в настоящее время является преувеличенной. Характерна высокая частота спонтанных мутаций гена TSC.
Сводные данные по TSC1 и TSC2 генам и спектру мутаций в них представлены в табл. 1. Скрининг мутаций является дорогостоящим и зависит от структуры и размера этих генов. Определенная патологическая мутация идентифицируется у 80–85 % больных. Некоторые исследования свидетельствуют о возможности сравнения спорадических случаев мутации в гене TSC1 и спорадических случаев мутации в гене TSC2. Так, в последнем случае имелась тенденция к более грубому фенотипу с высокой частотой умственной отсталости и судорог, более распространенным поражением почек и более выраженными ангиофибромами на лице. С другой стороны, некоторые миссенс-мутации в гене TSC2 могут передаваться и ассоциироваться с умеренной клинической картиной заболевания. В дополнение к генетической изменчивости разная тяжесть приписывается отличию между мутациями в TSC1 и TSC2 генах — они всегда поразительно изменчивы в выражении в пределах семьи. Это отчасти соответствует случайной природе соматической мутации, но сообщается также об ассоциации между высокой экспрессивностью аллеля в гене гамма-интерферона и низкой частотой почечных ангиомиолипом у пациентов с мутацией в гене TSC2. Также гены-модификаторы могут оказывать влияние на фенотип.
Соматический мозаицизм встречается у меньшинства пациентов с ТС, имеется склонность к ассоциации с «мягким» фенотипом. В этом случае мутации гена TSС присутствуют в низком проценте и трудны для нахождения, но соматический мозаицизм, несомненно, заслуживает внимания, когда мутации не поддаются идентификации. Герминативный мозаицизм — также давно признанный феномен ТС.
Продукты генов TSC1 и TSC2 — гамартин и туберин (димерные формы) являются ключевыми посредниками в фосфоинозитид-3-киназной сигнальной системе. Связывание внеклеточных ростовых факторов, в том числе инсулина, с рецепторами на клеточной мембране проходит через ряд шагов к фосфорилированию туберина. Это уменьшает ингибиторную активность гамартин-туберинового комплекса так, что он приводит в действие через малые GTPase Rheb (сервис удаленного доступа многостимульного гомолога в мозге) на мишени для рапамицина у млекопитающих (mTOR). Результатом активации mTOR является последовательное фосфорилирование двух из соответствующих мишеней — рибосомальной S6 киназы (S6K) и эукариотического инициаторного фактора 4Е связанного протеина 1 (4Е-BP1), обе из которых ведут к возрастанию переноса белка. Другие сигнальные системы оказывают влияние на активность гамартин-туберинового комплекса, который играет ключевую роль в суммировании информации о ростовой стимуляции, уровне клеточной энергии, питательных веществ и гипоксии и через mTOR регулирует синтез белка и клеточный рост. Функции гамартина и туберина самостоятельны и говорят об очевидности их связи с другими белками, но вызывает сомнения, которое из двух взаимодействий имеет какую-нибудь физиологическую значимость.
В настоящее время известно, что при ТС эктодерма поражается под влиянием генетических факторов еще в раннем эмбриональном периоде. Одновременно с поражением эктодермы появляются изменения и в других зародышевых листках.
Особенности клинической симптоматики
Манифестные формы ТС отличаются выраженным полиморфизмом и возрастзависимым дебютом. Единым гистологическим субстратом всех новообразований при ТС являются гамартомы. Гамартома (от греч. hamarta — ошибка) — опухоль из незрелой эмбриональной ткани, задержавшейся в своей дифференцировке по сравнению с окружающими тканями носителя опухоли, развивающейся из избыточно непропорционально развитых тканевых комплексов. Наиболее ярко выраженными и легко определяемыми клинически симптомами ТС являются кожные изменения, которые встречаются в 100 % случаев, и поражение центральной нервной системы (ЦНС).
Гипопигментированные пятна (гипомеланозные макулы) располагаются диффузно, асимметрично, преимущественно на животе, спине, передних и боковых поверхностях рук и ног (на кистях, стопах и гениталиях их не бывает). Они могут появляться с рождения или в более позднем возрасте, обычно в первые 2–3 года жизни. С возрастом наблюдается тенденция к увеличению их числа. Гипопигментированные пятна при ТС локализуются преимущественно на туловище и ягодицах. Характерной их особенностью является асимметричность расположения. Отмечена вариабельность числа, размера и формы пятен. Гипопигментированные пятна встречаются в 90 % случаев ТС и являются одной из наиболее характерных форм кожных проявлений. Гистологические исследования установили, что гипопигментированные пятна содержат нормальное число меланоцитов, но последние продуцируют меньшее количество слабо пигментированных меланосом. Цвет пятен матово-белый (в отличие от молочно-белой окраски при витилиго). Форма овальная или листовидная. Число пятен колеблется от 3–4 до 100 и более (симптом конфетти). Еще одним ярким клиническим симптомом заболевания являются депигментированные волосы, брови и ресницы.
Гиперпигментированные плоские пятна цвета кофе с молоком ( cafe-au-lait , франц.; milk coffee , англ.) и «веснушчатые гроздья» на коже, типичные для нейрофиброматоза, в 14,5 % случаев могут определяться и при ТС, но в последнем случае они встречаются реже, их число обычно не более пяти, что не превышает средних популяционных значений.
Аденомы сальных желез ( adenoma sebaceum , лат.), или, правильнее, ангиофибромы лица (аденома Прингла), — облигатный признак ТС, наблюдаются в 47–90 % случаев и появляются, как правило, после 4 лет жизни. Это особые узелковые высыпания на коже типа «просяных зерен», а иногда в виде массивных новообразований типа «цветной капусты». Внешний вид ангиофибром напоминает пятна или узелки с гладкой блестящей поверхностью. Они могут быть одиночными и множественными, плотны на ощупь, бледно-желтоватого, розово-красноватого цвета, наиболее часто располагаются в области носогубного треугольника, крыльев носа, на щеках и подбородке. Характерно их билатерально-симметричное расположение, что позволило сравнить ангиофибромы на лице с «крыльями бабочки». Ангиофибромы часто сопровождаются телеангиэктазиями, усиливающими красный цвет их окраски. По данным гистологии, эти образования состоят из гиперплазированных сосудов, разросшейся фиброзной ткани, незрелых волосяных фолликулов. Частота встречаемости — до 1/3 всех случаев ТС. Чаще дебютируют в возрасте 3–11 лет, в среднем — в 4–7 лет.
Участки «шагреневой кожи» ( peau chagrin , франц. — недубленая, грубая, жесткая кожа) — это четко выраженное локальное скопление фиброзных гамартом, сливающихся в относительно большое пятно. Пятна слегка выступают над поверхностью кожи, имеют желто-коричневую или розовую окраску, внешне напоминают свиную кожу или апельсиновую кожуру. Эти участки обычно располагаются на задней поверхности туловища, особенно часто — в пояснично-крестцовой области. Отличительной чертой является их асимметричность. Размер вариабелен: от нескольких миллиметров до 10 см и более. Количество участков «шагреневой кожи» вариабельно, но чаще они бывают единичными. Не являясь абсолютно патогномоничным симптомом ТС, тем не менее «шагреневые пятна» являются вторым по частоте встречаемости кожным симптомом после депигментированных пятен. Некоторые авторы считают участки «шагреневой кожи» облигатным признаком ТС и указывают частоту встречаемости до 21–68 % случаев. В большинстве случаев «шагреневая кожа» появляется на втором десятилетии жизни. Но поскольку фиброзные гамартомы имеют различные сроки дебюта, то их диагностическое значение уступает пигментным изменениям кожи при ТС.
Фиброзные бляшки — патогномоничный кожный симптом ТС, встречаются у 25 % больных. Фиброзные бляшки имеют бежевый цвет, шероховаты на ощупь и несколько выступают над окружающей кожей. Они часто появляются уже на первом году жизни и являются одним из первых клинических симптомов ТС. Однако чаще фиброзные бляшки появляются в более зрелом возрасте по сравнению с ангиофибромами лица. Как правило, бляшки располагаются на лбу и волосистой части головы унилатерально. Бляшки имеют разную консистенцию, выступают над поверхностью кожи, шероховатые на ощупь. Их размеры и число варьируют.
Мягкие фибромы встречаются у 30 % больных. Это множественные или единичные мягкие образования на ножках, мешотчатой формы, растущие на шее, туловище и конечностях ( molluscum fibrosum pendulum ). Другой вариант мягких фибром представляет собой множественные, несколько приподнятые над поверхностью кожи (и такого же цвета) мелкие образования, размером меньше булавочной головки, располагающиеся на туловище и шее и напоминающие гусиную кожу.
Околоногтевые фибромы (опухоли Коенена) — тусклые, красные или цвета кожи узелки, расположенные на пальцах, или на латеральной поверхности ногтевого ложа под ногтевой пластинкой, или вдоль проксимального ногтевого валика. Чаще встречаются на пальцах стоп, преимущественно у женщин. Размер фибром варьирует от 1 до 10 мм. Околоногтевые фибромы являются возрастзависимым клиническим симптомом ТС (обычно появляются во время или непосредственно после пубертата) и могут встречаться у 50 % больных. Эти образования склонны к прогрессивному росту даже после их удаления.
Поражение ЦНС характерно для ТС. Наиболее типичным поражением являются корковые туберсы и субэпендимальные узлы . Макроскопически на выпуклой поверхности больших полушарий головного мозга по ходу извилин выявляется множество плотных образований (туберсов) желто-белого цвета, в центре которых часто имеются рубцовые втяжения. Туберсы также могут располагаться перивентрикулярно, часто вдаваясь в просвет боковых желудочков (субэпендимальные узлы), иногда приводя к развитию внутренней окклюзионной гидроцефалии. Туберсы следует рассматривать как гамартомы (неправильно сформировавшиеся эмбриональные тканевые комплексы опухолевого генеза без видимых признаков прогрессирующего роста). Они могут быть как единичными, так и множественными, располагаются в виде выступов над единичной или прилегающими бороздами коры, расширяя их. Строение перивентрикулярных туберсов идентично строению корковых, но в них нередко образуются гемангиоматозные структуры, и они часто подвергаются обызвествлению. В целом кальцификация туберсов отмечается в 54 % случаев и увеличивается с возрастом больных. Своевременное выявление корковых туберсов и кальцификатов мозга очень важно для диагностики TС.
Субэпендимальные узлы встречаются в 95 % случаев и выявляются как при КТ, так и при МРТ мозга. В большинстве случаев они множественные, прилегающие друг к другу, локализуются в стенках боковых желудочков и, реже, в стенках III и IV желудочков мозга. При локализации в стенках боковых желудочков субэпендимальные узлы глубинной частью могут внедряться в хвостатое ядро или таламус. Их форма обычно округлая или вытянутая. У новорожденных субэпендимальные узлы редко бывают кальцифицированными. По мере роста ребенка наблюдается постепенное отложение кальция в субэпендимальных узлах. Субэпендимальные узлы в 10–15 % случаев трансформируются в гигантоклеточную астроцитому, которая манифестирует обычно между 5-м и 10-м годами жизни больного, имеет тенденцию к росту и локализуется у межжелудочкового отверстия Монро. Для диагностики гигантоклеточных астроцитом применяется как КТ, так и МРТ. В клинической практике нередко используют оба нейрорадиологических метода визуализации. Учитывая тот факт, что при ТС гигантоклеточные астроцитомы наблюдаются относительно часто, рекомендуется динамическое проведение КТ/МРТ у больных детей (не реже 1 раза в 2 года). В случае появления таких клинических симптомов, как головная боль, рвота, ухудшение зрения, необходимо экстренное проведение нейрорадиологического исследования.
Помимо субэпендимальных узлов и кортикальных (или внутримозговых) туберсов могут быть выявлены и другие типы церебральных изменений, характерных для ТС. Например, субэпендимальные гигантоклеточные образования (СГО). Относительную редкость обнаружения такого рода изменений у детей с ТС можно объяснить их медленным ростом — пика визуализации они достигают при возрасте пациента более 11–13 лет. Основное отличие СГО от субэпендимальных узлов состоит не в их морфологической структуре, a в размерах (они всегда существенно превышают размеры субэпендимальных узлов) и, самое главное, в тенденции к постепенному увеличению. Можно предположить, что СГО — это, по сути, те же субэпендимальные узлы, клетки которых по неясной причине не утратили способности к репродукции. Считается, что СГО выявляются примерно в 10–15 % случаев ТС. Как и субэпендимальные узлы, СГО локализуются преимущественно вокруг отверстия Монро, при этом часто деформируя его и приводя к разбитию вторичной внутренней обструктивной гидроцефалии.
Также наблюдаются участки фокальной корковой дисплазии (ФКД), которые проявляются ограниченной дисплазией кортикальной пластинки в виде ее утолщения, стушеванности границы между серым и белым веществом, изменения рисунка прилегающих борозд и формы пораженной извилины. Гигантские нейроны и причудливой формы гигантские астроциты, которые считаются патоморфологической основой синдрома ФКД, характерны и для туберозного склероза. Идея относительно гистопатологического родства ФКД и туберозного склероза получила максимальное развитие в работе R. Bronen и соавт., которые предположили, что сама по себе типичная баллоноклеточная ФКД («тэйлоровский» тип) является вариантом «фруст-формы» ТС и ее нужно рассматривать как результат неполной пенетрантности гена TSС. Кроме того, родоначальник учения о ФКД Дэвид Тэйлор высказывал мнение о ФКД как о не совсем типичном проявлении внутримозговых изменений при ТС. Не случайно для обозначения ФКД он какое-то время использовал ныне забытый термин «тубероидный псевдосклероз».
При ТС могут быть и изменения белого вещества, которые по МРТ-характеристикам соответствуют островкам гетеротопированных нейронов, ассоциированных с участками гипомиелинизации. Поражение белого вещества проявляется изо- или гипоинтенсивностью на Т1-взвешенных срезах и абсолютной гиперинтенсивностью на Т2-взвешенных срезах. Можно выделить три основных топографических паттерна локализации очагов в белом веществе при ТС: прямые или изогнутые лентовидные участки, которые распространяются от стенки желудочка трансцеребрально вплоть до кортикальной поверхности; клиновидные участки разной протяженности; неспецифические по форме конгломеративные фокусы измененной интенсивности МР-сигнала. Особый интерес представляют лентовидные трансцеребральные фрагменты, так как их форма и расположение наталкивают на мысль, не являются ли они «остановившейся во времени» миграционной дорожкой.
Поражения ЦНС являются доминирующими в клинической картине ТС. Классическая клиническая картина включает: эпилептические приступы, задержку умственного развития, нарушения поведения, изменения в цикле «сон — бодрствование». Исключение составляют «фруст-формы» (стертые формы), при которых наряду с наличием типичных нейрорадиологических проявлений и кожных изменений могут отсутствовать эпилептические приступы, умственная отсталость или нарушение поведения. Эпилептические приступы — один из наиболее значимых симптомов ТС. Они наблюдаются у 80–92 % больных и чаще всего являются манифестным симптомом заболевания. Эпилептические приступы при ТС нередко резистентны к противоэпилептической терапии, могут приводить к развитию нарушений интеллекта и поведения и являются одной из главных причин инвалидности у детей. P. Curatolo отмечено, что среди факторов, детерминирующих резистентность к противоэпилептической терапии, наибольшее значение имеют: дебют в возрасте до 1 года, наличие нескольких типов приступов, высокая частота приступов, изменение характера приступов с течением заболевания. Эпилептические приступы при ТС дебютируют на первом году жизни. Чаще всего в этом возрастном периоде встречаются инфантильные спазмы (синдром Веста), которые в дальнейшем переходят в другие формы фокальной эпилепсии и эпилептических синдромов (например, синдром Леннокса — Гасто). В большинстве случаев наблюдаются инфантильные спазмы, атипичные абсансы, фокальные или комплексные психомоторные и соматомоторные припадки (с джексоновским маршем или без такового), возможны вторично-генерализованные тонико-клонические припадки. В единичных наблюдениях эпилептические приступы, возникая с первого года жизни ребенка, являются первыми клиническими симптомами ТС. В большинстве случаев эпилептические припадки являются фармакорезистентными.
Умственная отсталость наблюдается у 50 % больных с ТС. Одна из главных причин — симптоматическая эпилепсия, дебютирующая на первом году жизни больного ребенка. Другая причина — корковые гамартомы (туберсы), локализующиеся в теменной, височной и лобной долях головного мозга. Число корковых туберсов у больных с ТС коррелирует с нарушениями обучения и проявлениями аутизма. Степень умственной отсталости колеблется от умеренной до глубокой. Изменения поведения характеризуются аутизмом, синдромом дефицита внимания с гиперактивностью, агрессией и/или аутоагрессией. Задержка в умственном и речевом развитии наблюдается более чем в половине случаев ТС. При этом с первых лет жизни обнаруживаются моторное возбуждение, общее беспокойство, которые напоминают полевое поведение при аутизме. Характерно нарастание психопатических черт в поведении с недовольством, капризностью, взрывчатостью, застреваемостью на аффективных реакциях. Дети медлительны, торпидны, с трудом переходят от одного вида деятельности к другому. В случаях выраженной задержки умственного развития коммуникативность у больных с ТС утрачивается. Моторное беспокойство сменяется малой подвижностью ребенка. Чем раньше дебютирует ТС, захватывая период от первых месяцев жизни до 1,6 года, тем грубее умственное недоразвитие, что соотносится с коморбидностью заболевания с критическим периодом нейроонтогенеза, явлениями физиологического апоптоза. При этом в 50 % случаев наблюдаются умственное недоразвитие и задержка развития локомоторно-статических функций (Л.М. Калинина, 1973). У этих детей нередко до 2–5 лет ставят диагноз аутизма или аутистических черт с тяжелой гиперактивностью (A. Hunt, J. Denis, I. Gillberg, 1987; Ch. Gillberg, G. Ahelsen, 1994).
Нарушения сна у большинства больных с ТС связаны с длительным периодом засыпания, бессонницей, частыми ночными пробуждениями, снохождением (сомнамбулизмом) и ранним пробуждением.
Глазные симптомы выявляются у 50 % больных, часто манифестируют в первые 2 года жизни и условно подразделяются на две большие группы: ретинальные и неретинальные. Наиболее часто встречаются ретинальные гамартомы (факомы) , обычно двусторонние и часто множественные. По внешнему виду эти опухоли подразделяются на кальцинированные (симптом тутовой ягоды) и некальцинированные. Они располагаются поверхностно по отношению к сосудам сетчатки и могут быть обнаружены, когда эти сосуды кажутся прерванными или частично затемненными. Факомы обычно локализованы около или по краю диска зрительного нерва, но могут быть выявлены и на периферии сетчатки. Их размеры варьируют от 0,5 до 4 диаметров диска зрительного нерва. В настоящее время выделяют 3 наиболее типичных варианта гамартом сетчатки. При первом, наиболее распространенном варианте гамартомы имеют нежную, относительно плоскую и гладкую поверхность, оранжево-розовый цвет, округлую или овальную форму, локализуются преимущественно в поверхностных слоях сетчатки. При втором — гамартомы имеют узловатый вид и напоминают тутовую ягоду. Они белого цвета, кальцифицированные, светонепроницаемые. При третьем варианте гамартомы сочетают в себе признаки первых двух. Они имеют округлую форму с узловатым и кальцифицированным центром и полупрозрачной, гладкой периферией оранжево-розового цвета. Клинические проявления гамартом наблюдаются крайне редко. Основным симптомом является прогрессирующее снижение зрения. Следует помнить, что в некоторых случаях ТС проявляется только специфическими офтальмологическими изменениями. Неретинальные глазные симптомы ТС включают ангиофибромы век, депигментацию радужной оболочки (пятна Лиша) или сетчатки, атипичную колобому, катаракту, отек диска зрительного нерва, выпадение полей зрения (гомонимную гемианопсию), непаралитическое косоглазие, парез VI нерва, кровоизлияния в стекловидное тело.
Изменения внутренних органов при ТС включают: рабдомиомы сердца, ангиомиолипомы почек, почечные кисты (поликистоз почек), почечноклеточную карциному, лимфоангиомиоматоз легких, ангиомиолипомы надпочечников, печени, ректальные полипы. Характерной особенностью этих изменений является их множественный характер, часто билатеральное расположение в парных органах, возможность длительного бессимптомного течения, а также возрастзависимая манифестация, которая колеблется в широком возрастном диапазоне — от 5–7 до 30–40 лет жизни.
Рабдомиома (фетальная гамартома) — патогномоничное для ТС поражение сердца. Вовлечение сердца отмечается часто, но обычно протекает бессимптомно. Кардиальные нарушения рабдомиомы вызывают сравнительно редко, но у больных более юного возраста они встречаются чаще, чем в более старших возрастных группах. Клинические симптомы рабдомиом у новорожденных различны. При опухолях, диагностированных внутриутробно, особенно при массивных опухолях, в 25–30 % случаев наблюдается внутриутробная смерть плода либо дети рождаются преждевременно с низкой оценкой по шкале Апгар, имеют распространенные отеки и выраженный цианоз. Имеются сообщения о случаях смерти новорожденных от застойной сердечной недостаточности. Примерно у 50 % новорожденных опухоль может выявиться случайно при проведении планового эхокардиографического обследования по поводу ТС. Обычно эти опухоли не нарушают гемодинамику и не имеют выраженного интрамурального роста. Известны случаи диагностики рабдомиомы при обследовании новорожденных по поводу пароксизмальной тахикардии. Рабдомиомы локализуются в любой полости сердца, но преимущественно в желудочках. Более характерно их расположение в левой половине сердца. В очень редких случаях рабдомиомы могут локализоваться в предсердиях, исходя из межпредсердной перегородки. Она может располагаться интрамурально или пролабировать в полость сердца. Кардиологические симптомы рабдомиомы у детей младшего возраста включают: нарушения сердечного ритма (тахикардия, миграция водителя ритма, блокада сердечной проводимости, синдром Вольфа — Паркинсона — Уайта, фибрилляция желудочков), нарушение сократительной функции миокарда при интрамуральном расположении опухоли, сердечную недостаточность вследствие обструкции камер сердца опухолью. У детей старшего возраста рабдомиомы сердца обычно бессимптомны. Редко могут наблюдаться преходящая блокада левой ножки пучка Гиса, псевдоишемические изменения на электрокардиограмме. Обычно рабдомиома не имеет капсулы (некапсулированная доброкачественная опухоль), но четко отграничена от окружающих тканей, может состоять из одного узла или быть множественной. Замечено, что рабдомиомы сердца у больных с ТС, как правило, быстро увеличиваются во время 2-й половины беременности, в основном достигают максимальных величин к моменту рождения, а затем постепенно уменьшаются в размерах. Большинство рабдомиом исчезает бесследно. Спонтанное регрессирование рабдомиом описано у детей младше 6 лет. После 6 лет опухоли обычно не исчезают, однако могут несколько уменьшаться в размере. Регрессирование опухолей при ТС может наблюдаться как в размере, так и в числе. Случаи малигнизации этой опухоли не описаны. Кардиальные нарушения могут приводить к летальному исходу даже в раннем детском возрасте.
Изменения со стороны легких. Легкие вовлекаются в патологический процесс у больных ТС после 30 лет. Первые клинические симптомы лимфангиомиоматоза легких, как правило, включают дыхательную недостаточность и рецидивирующий пневмоторакс. На рентгенограмме грудной клетки выявляется усиленный рисунок легочной паренхимы и характерный паттерн «сотовых» легких (кисты легких), распространяющийся на всю их паренхиму или только на ее изолированные участки.
Изменения со стороны желудочно-кишечного тракта у больных с ТС многообразны, в патологический процесс могут быть вовлечены: полость рта (узловые опухоли, фибромы или папилломы, дефекты эмали зубов); печень (одиночные и множественные гамартомы и ангиомиолипомы); прямая кишка (ректальные полипы). Наиболее типичные нарушения, выявляемые при исследовании ротовой полости, — это узловые опухоли, фибромы или папилломы. Они локализуются главным образом на переднем крае десен, преимущественно на верхней челюсти, но также встречаются на губах, слизистой оболочке щек, спинке языка и небе. Дефекты эмали зубов отмечаются практически у всех больных. Возможны несколько вариантов дефектов эмали зубов: небольшие углубления, невидимые без увеличения, около 4 мкм в диаметре; углубления в эмали зубов до 60 мкм в диаметре; кратерообразные углубления в эмали зубов, видимые невооруженным глазом, около 100 мкм в диаметре. Одним из наиболее типичных нарушений является дефект эмали зубов в виде углублений, число которых варьирует от 1 до 11 на каждом зубе.
Полипы прямой кишки, которые встречаются в 50–78 % случаев ТС, протекают бессимптомно (лишь в отдельных случаях возможны боли при дефекации) и не склонны к малигнизации. Обычно выявляются у больных старше 20 лет, диагностируются при пальцевом исследования прямой кишки и/или с помощью инструментальных методов исследования (сигмоидоскопии, колоноскопии, контрастной рентгенографии прямой кишки). Как правило, ректальные полипы имеют благоприятный прогноз.
Изменения со стороны почек встречаются в 47–85 % случаев ТС, причем изолированное поражение почек в дебюте ТС встречается в 1–2 %, а при аутопсийном исследовании — в 100 % случаев. При этом ангиомиолипомы выявляются у 48 % больных, кисты — у 35 %, а сочетание ангиомиолипом и кист — у 17 %. Реже встречаются почечноклеточная карцинома (в 5 %), онкоцитома, а также неопухолевое поражение почек — фокальный сегментарный гломерулосклероз. Описаны также сосудистые дисплазии и пороки почечной ткани, тубулоинтерстициальный нефрит, мембранопролиферативный гломерулонефрит и нефрокальциноз. Как правило, поражение почек при ТС манифестирует во 2–3-й декадах жизни, но клинические симптомы могут проявляться и в более ранние сроки.
Ангиомиолипомы почек выявляются у 2/3 взрослых больных ТС. Обычно ангиомиолипомы множественные, двусторонние и длительное время имеют бессимптомное течение. По данным Steiner и соавт., у 17 % больных с ангиомиолипомами почек выявляются признаки ТС, причем нередко отмечается двустороннее поражение. Хотя в небольшой части случаев ангиомиолипомы имеют склонность к малигнизации, они протекают асимптомно значительно чаще, чем кисты. При УЗИ почек ангиомиолипомы выглядят как эхопозитивные округлые образования в почечной паренхиме. При величине их менее 4 см в диаметре, как правило, никаких клинических проявлений не отмечается. При дальнейшем росте в центре ангиомиолипомы может появиться участок просветления, свидетельствующий о потенциальной возможности забрюшинного или внутрипочечного кровотечения. Считается, что ангиомиолипомы больше 4 см в диаметре имеют тенденцию к спонтанному кровоизлиянию. Кровотечение может быть как острым, так и хроническим и квалифицируется как угрожающее жизни состояние. Основными клиническими симптомами кровоизлияния из ангиомиолипомы являются: острые абдоминальные боли, резкое снижение артериального давления, холодный пот, анемия, возможна гематурия и микроальбуминурия. Эти опухоли в редких случаях сдавливают почечные артерии, приводя к гипертензии. Иногда они вызывают боли и кровотечения, а в части случаев ― хроническую почечную недостаточность (ХПН).
Кисты почек у больных с ТС могут развиваться в любом отделе нефрона, бывают единичными или множественными, часто неотличимыми от кист при поликистозной болезни взрослого типа. Чаще кисты почек небольшого размера, двусторонние. Они обнаруживаются более чем в 1/3 случаев ТС, но клинические проявления они вызывают лишь у небольшого числа больных. Патогномоничный морфологический признак ТС — внутренняя выстилка кист крупными клетками с эозинофильной цитоплазмой, образующими гиперпластические узелки, которые могут заполнять полость кисты. Почечные кисты выявляются при мутации как гена TSC1, так и гена TSC2. При мутации гена TSC1 кисты чаще солитарные, односторонние и выявляются реже, чем при мутации гена TSC2. Возраст выявления кист различен. Наиболее часто они диагностируются позже 10 лет, однако в ряде случаев поликистоз выявляется у детей раннего возраста и даже при ультразвуковом исследовании плода. Первыми клиническими симптомами кистоза почек могут быть боль в пояснице или гематурия. Исключением является рано выявленная поликистозная болезнь, которая может дебютировать артериальной гипертензией, обусловленной активацией ренин-ангиотензин-альдостероновой системы. Для постановки диагноза кистоза почек достаточно ультразвукового исследования. Внутривенная урография проводится в тех случаях, когда имеет место деформация чашечно-лоханочной системы с нарушением оттока мочи, присоединение инфекции. Часто поликистоз осложняется артериальной гипертензией, инфекцией мочевых путей, реже — кровотечением. Снижение функции почек, как правило, выявляется в 3–4-й декадах жизни. Прогрессированию почечной недостаточности способствуют тяжелые физические нагрузки, а также беременность. Однако в целом при ТС терминальная ХПН возникает редко.
В возрастном контингенте 20–30 лет к другой по частоте патологии почек при ТС относятся гамартомы, которые могут сочетаться с саркомой почки. Злокачественные опухоли при ТС выявляются в 4,4 % случаев, то есть чаще, чем в популяции, с преобладанием у женщин (81 %). Средний возраст их выявления — 29 лет. Билатеральное поражение встречается в 25 % случаев. Наиболее часто диагностируется почечноклеточная карцинома.
Летальность, связанная с патологией почек при ТС, занимает 2-е место в структуре смертности после поражения ЦНС. Наиболее часто причиной смерти становится хроническая почечная недостаточность. В 1996 г. F. Shillinger и R. Montagnac описали 65 случаев ТС, все больные наблюдались в центрах гемодиализа Франции. По данным авторов, частота терминальной хронической почечной недостаточности составила 1 на 100 случаев заболевания. Чаще она выявлялась у женщин (63,1 %) и диагностировалась в среднем в 29-летнем возрасте. В половине случаев хронической почечной недостаточности она была первым симптомом манифестации ТС. Причинами ХПН были: ангиомиолипоматоз почек (23 %), поликистоз почек (18,5 %), сочетание кист и ангиомиолипом почек (53 %), злокачественные опухоли почек (13,8 %). ХПН при ТС связана со сдавлением функционирующей ткани опухолевыми массами и/или кистами, а также с частичной или тотальной нефрэктомией (при операциях по поводу осложненных ангиомиолипом), что ведет к гиперфильтрации в сохранных нефронах и возникновению вследствие этого гломерулосклероза.
Костные изменения являются случайной находкой у больных с ТС, они часто не проявляют себя клинически и выявляются при плановых рентгенологических исследованиях. Наблюдаются кистоподобные изменения и склеротические участки, излюбленной локализацией которых являются кости свода черепа, таза и позвонки, утолщение коркового слоя трубчатых костей, дефекты эмали зубов.
Заключение
Несмотря на невысокую частоту встречаемости ТС (болезни Бурневилля — Прингла) в популяции, тяжелая инвалидизация и отсутствие эффективных средств лечения придают данной проблеме исключительную актуальность. Неуклонный рост данной патологии в структуре общей заболеваемости можно объяснить поздней диагностикой, несвоевременной профилактикой и ухудшением экологической обстановки. Своевременно поставленный диагноз позволяет определить дальнейшую тактику ведения больного, а также обеспечить медико-генетическое консультирование членам семьи, что должно способствовать снижению частоты рождения больных с тяжелыми формами заболевания. Первичная диагностика ТС должна осуществляться педиатрами, подростковыми врачами, участковыми врачами и врачами общей практики (семейными врачами), а также узкими специалистами (неврологами, дерматологами, офтальмологами, хирургами, стоматологами) в процессе динамического диспансерного обслуживания населения. Важно помнить, что процесс развития клинической симптоматики ТС является динамическим, поэтому важна преемственность между специалистами различного профиля и своевременное проведение комплекса дополнительных методов диагностики, включая современные методы визуализации — КТ/МРТ головного мозга и органов брюшной полости.
Продолжение.
Продолжение.
Кириллова Л.Г., Шевченко А.А., Лисица В.В., Силаева Л.Ю., ГУ «Институт педиатрии, акушерства и гинекологии АМН Украины»
Согласно классификации мальформаций развития коры, туберозный склероз относится к группе врожденных пороков, возникающих вследствие нарушений нейрональной пролиферации и дифференциации. В 3–4 месяца внутриутробного развития происходит закладка и других тканей организма. Следовательно, воздействие патогенного фактора или реализация генной мутации на этом этапе вызывает повреждение нескольких органов, что может быть определено при ультразвуковом обследовании беременной. Пренатально заподозренная патология позволяет избрать правильную тактику ведения родов, организовать пристальное наблюдение за состоянием ребенка с первых дней его жизни и своевременно назначить необходимую терапию.
Туберозный склероз (болезнь Бурневилля) является редким генетическим заболеванием, характеризующимся полисистемностью поражения. Туберозный склероз был впервые описан в 1862 году немецким патологом Фридрихом фон Реклингхаузеном, работающим в качестве ассистента профессора Рудольфа Вирхова в Институте патологической анатомии в Берлине. Реклингхаузен представил сердце умершего вскоре после рождения младенца с несколькими опухолями и назвал их миомами. У этого же ребенка был отмечен кортикальный «склероз» мозга. По всей видимости, это были сердечные рабдомиомы и кортикальные туберсы. В 1880 году известный французский невролог Дезире-Маглуар Бурневилль детализировал выявленные патологические данные и впервые применил термин «туберозный склероз». Исследуя заболевание молодого человека, характеризующееся эпиприпадками, гемиплегией, задержкой умственного развития, а также наличием почечных опухолей, Бурневилль определил данную патологию как «туберозный склероз церебральных волокон». У этого же пациента впервые была описана сыпь на носу, щеках и лбу, появившаяся в подростковом возрасте [9, 29].
В результате многолетних исследований в 1998 году Согласительной комиссией по туберозному склерозу были приняты диагностические критерии данного заболевания. Для постановки диагноза необходимо наличие 3 признаков: полиморфных припадков, резистентных к медикаментозной терапии, изменений со стороны кожи и прогрессирующей задержки психоречевого и двигательного развития [2, 9].
В настоящее время туберозный склероз, или болезнь Бурневилля, относят к генетически детерминированным заболеваниям. Отмечается два варианта поражения генов. Первый вариант туберозного склероза развивается при мутации гена, расположенного в хромосоме 9q34 — в участке 34 длинного плеча хромосомы 9 (туберозный склероз 1-го типа — TSC1, кодирует белок гамартин, открыт в 1987 году), второй — в связи с мутацией другого гена на хромосоме 16р13 — в участке 13 короткого плеча хромосомы 16 (туберозный склероз 2-го типа — TSC2, кодирует белок туберин, открыт в 1992 году) [11, 38]. Известно, что мутация перечисленных генов является основой для развития гиперпластических процессов в связи со снижением синтеза белков гамартина и туберина, которые в норме подавляют опухолевый рост в организме и недостаточно вырабатываются у больных с туберозным склерозом. Мозаицизм туберозного склероза встречается тогда, когда только часть клеток организма пациента содержит мутации в генах TSC1 или TSC2. Пациенты с мозаичным генотипом могут иметь полный спектр симптомов туберозного склероза [31]. Существуют исследования по сравнению спорадических случаев мутации в гене TSC1 и спорадических случаев мутации в гене TSC2. В частности, в последнем случае отмечалась тенденция к более грубому фенотипу с высокой частотой умственной отсталости и судорог, более распространенным поражением почек и более выраженными ангиофибромами на лице. С другой стороны, некоторые миссенс-мутации в гене TSC2 могут передаваться и ассоциироваться с умеренной клинической картиной заболевания [40].
Туберозный склероз наследуется по аутосомно-доминантному типу с варьирующей экспрессивностью и почти 100% пенетрантностью. Как спонтанная мутация заболевание встречается в 60–70 % случаев (ни один из родителей не имеет данной патологии); при наличии туберозного склероза у одного из родителей вероятность его возникновения у ребенка составляет 50 % [5, 30, 33].
Трудно судить о частоте заболевания туберозным склерозом, так как в клиниках основным диагнозом зачастую выступает симптоматическая эпилепсия или задержка развития. Согласно оценкам экспертов, туберозный склероз отмечается у 2 миллионов человек во всем мире, с одинаковой частотой во всех расах [5, 30, 33, 39].
Она выше у детей, составляя максимум у не достигших 5-летнего возраста. Границы диапазона распространенности заболевания в детстве варьируют от 1 на 6800 до 1 на 17 300 [39].
Среди наиболее распространенных причин смерти у больных с туберозным склерозом следует выделить эпилептический статус, бронхопневмонию и почечную недостаточность [34].
Данная патология характеризуется бугорковоподобными разрастаниями в веществе головного мозга, а также в коже, почках, глазах, легких, сердце, костях. Вероятной причиной опухолеобразования считается трансформация ряда клеточных элементов в так называемые РЕС-клетки (эпителиоидные мышечные клетки), характерной чертой которых являются нарушения митохондриального аппарата [24]. Характерные новообразования и изменения кожи лица, туловища, конечностей и глазного дна могут быть учтены при начальной диагностике [35].
Классической клинической триадой туберозного склероза являются припадки судорог, умственная отсталость и специфическая сыпь на лице [30, 33].
Частота эпилептических припадков при туберозном склерозе варьирует от 70 до 90 %. Встречаются разные типы эпиприпадков, и выявлена их определенная зависимость от возраста больных. Например, инфантильные спазмы являются более характерными для детей первого года жизни, парциальные припадки теменно-затылочной локализации возникают у детей до 2-летнего возраста, а позднее возникают припадки с локализацией очага в височной и лобной областях. У большинства пациентов с генерализованными припадками данные ЭЭГ и видеомониторинга выявляют фокальное или мультифокальное начало припадков. У многих больных наблюдается корреляция электроэнцефалографического очага с мальформациями коры (бугорками), выявляемыми при нейровизуализации [3, 5, 7, 23, 33].
Некоторыми авторами отмечено, что эпиприпадки встречаются у всех больных с нарушением интеллекта и у 70 % пациентов без нарушений [2, 8, 9]. Отличительной чертой эпилептических припадков при туберозном склерозе является резистентность. Именно поэтому во время Международного симпозиума «Резистентные формы эпилепсий и эпилептические энцефалопатии у детей», проходившего в Донецке 30–31 октября 2008 года, доклад Паоло Куратоло, который в настоящее время является одним из экспертов по вопросам туберозного склероза в Европе, был посвящен анализу особенностей эпиприпадков при данной патологии. Профессор П. Куратоло отметил, что более чем у 65 % пациентов эпилептические припадки при туберозном склерозе начинаются в возрасте до одного года, при этом треть случаев представлена инфантильными спазмами, а в 60 % случаев формируется фармакорезистентность. Парциальные приступы чаще возникают в дебюте заболевания и с течением времени трансформируются или наблюдаются параллельно со спазмами. В возрасте 2–5 лет в клинической картине могут появляться сложные парциальные припадки и дроп-атаки. Для лечения в качестве препарата первой линии выбора используется вигабатрин в дозе 50–100 мг/кг в сутки, а другими эффективными препаратами могут служить леветирацетам и топирамат. Обращено внимание, что более чем у половины больных имеют место и другие изменения в виде снижения интеллекта — в 55 % случаев, синдрома дефицита внимания и гиперактивности — в 30 % , аутизма — в 28 % [3].
Умственная отсталость различной степени выраженности выступает вторым по частоте клиническим проявлением при туберозном склерозе, отмечается у 50–85 % пациентов. В 2/3 случаев интеллектуальная недостаточность является глубокой (IQ < 21), у 50 % наблюдаются трудности в обучении [27]. Одними из основных причин умственной отсталости являются судороги, возникающие на первом году жизни, и недостаточный медикаментозный контроль над эпилептическими припадками [39]. Еще одна причина — наличие корковых гамартом (туберсов) с локализацией в теменной, височной и лобной долях головного мозга. Также в ряде публикаций отмечено, что количество корковых туберсов у пациентов с данной патологией связано с нарушениями обучения и проявлениями аутизма [27, 30].
Необходимо подчеркнуть, что гипопигментированные пятна на туловище и конечностях являются одним из первых проявлений туберозного склероза и выявляются даже у новорожденных, хотя могут быть ложно диагностированы как родимые пятна. Они лучше визуализируются в ультрафиолетовых лучах и встречаются в 83 % случаев [5, 30]. Гипопигментация обычно представлена многочисленными пятнами преимущественно на животе, спине, передних и боковых поверхностях рук и ног. На более поздних этапах характерными являются ангиофибромы лица (на щеках, подбородке, около носа), что встречается в 50–80 % случаев с проявлениями в виде сыпи, состоящей из пятен размером 0,1–1 см. При распространении по лицу они могут принимать фигуру «бабочки». Со временем пятна имеют тенденцию к слиянию и изменению цвета (коричневый, темно-красный). В более старшем возрасте в 30–70 % случаев диагностируются участки «шагреневой кожи», подногтевые фибромы, диффузное повреждение эмали зубов [3, 20].
Доброкачественные новообразования — липомы, фибромы, аденомы и т.п. — могут встречаться практически во всех внутренних органах [5].
Примерно в 50 % случаев у больных развиваются рабдомиомы, которые могут быть обнаружены с помощью эхокардиографии [9]. Эти доброкачественные опухоли располагаются в любой сердечной полости, но чаще они встречаются в желудочках. Рабдомиомы часто развиваются в сроке 22–26 недель беременности и могут вызывать клапанную дисфункцию, затруднение оттока, уменьшение сократимости миокарда, кардиомиопатию, а в некоторых случаях гибель эмбриона. Рабдомиомы вызывают кардиальные нарушения сравнительно редко и у больных более юного возраста. Осложнения могут приводить к летальному исходу даже в раннем детском возрасте. Хирургическому лечению подлежат только внутриполостные рабдомиомы, приводящие к гемодинамической обструкции [15, 16, 33]. В связи с этим следует акцентировать внимание на значимости пренатальной ультразвуковой диагностики, позволяющей обнаружить данную опухоль.
Исследования некоторых зарубежных авторов в 90-х годах ХХ века, проводивших МРТ мозга плодов, у которых при УЗИ выявлены изменения сердца, позволили пренатально подтвердить наличие субэпендимальных узлов в желудочках мозга почти у половины обследованных. Авторы считают, что наличие более одного субэпендимального узла (под эпендимой боковых желудочков) не вызывает сомнения в диагнозе туберозного склероза [9, 15, 16].
С частотой до 70–80 % при туберозном склерозе отмечается поражение почек в виде 2 основных патологических образований: ангиомиолипомы и кисты [26]. Кисты обнаруживаются более чем в трети случаев туберозного склероза, но клинические проявления они вызывают лишь у небольшого числа больных и более характерны для детей. Ангиомиолипомы в почках выявляются у 2/3 взрослых пациентов. В небольшой части случаев ангиомиолипомы имеют склонность к малигнизации, но протекают асимптомно значительно чаще, чем кисты, и только в редких случаях сдавливают почечные артерии, что приводит к гипертензии, иногда вызывают боли и кровотечения, а в некоторых случаях почечную недостаточность [21, 22, 26].
Также у многих больных отмечаются полипы прямой кишки, которые протекают бессимптомно и не склонны к малигнизации [33]. Кистозное поражение легких наблюдается редко и выявляется обычно к 30–40-летнему возрасту, но, прогрессируя, может приводить к смерти больных [14]. Исследования печени у детей с туберозным склерозом обнаружили гамартомы у 23,5 % пациентов [17].
Примерно у половины больных туберозным склерозом имеет место патология глаз в виде астроцитарных гамартом сетчатки. На глазном дне отмечаются классические рельефные изменения в виде тутовой ягоды, но пораженная полупрозрачная плоскость весьма трудна для идентификации. Гамартомы сетчатки редко нарушают зрение, но могут использоваться как дополнительный диагностический знак [33, 40].
Поражения нервной системы являются особенно существенными в прогнозе туберозного склероза. Более чем у 95 % пациентов могут отмечаться следующие, представленные ниже структурные аномалии мозга:
— кортикальные/субкортикальные туберсы;
— субэпендимальные узлы;
— субэпендимальные гигантоклеточные астроцитомы;
— «кистоподобные» поражения;
— агенезия/дисплазия мозолистого тела;
— связь с гемимегаленцефалией, шизенцефалией, внутричерепными артериальными аневризмами достигает 60 % [9, 19].
В гистологическом плане бугорковоподобные разрастания (бугорки) в мозге при туберозном склерозе являются участками дезорганизации коры с отсутствием ее нормальной многослойной архитектоники. Субэпендимальные узелки содержат в себе смесь сосудистой стромы и астроцитоподобных клеток, некоторые из которых являются подобными большим клеткам в бугорках. Бугорки могут быть ассоциированы с аномалиями нижележащего белого вещества в результате нарушения миграции нейронов. Субэпендимальные узелки обычно остаются скрытыми, бессимптомными на всем протяжении жизни, но имеют потенциал увеличения в размерах и развиваются в субэпендимальную гигантоклеточную астроцитому. Сама же субэпендимальная гигантоклеточная астроцитома встречается в 6–14 % случаев туберозного склероза с пиком охвата в позднем детстве и подростковом возрасте [9, 40].
Узлы от 1–2 мм до нескольких сантиметров в диаметре локализуются чаще в извилинах большого мозга, в эпендиме желудочков. В 30–80 % случаев возможно их обызвествление. Расположение кальцификатов в кортексе и около желудочков является наиболее характерным для туберозного склероза [4]. Также были отмечены участки фокальной корковой дисплазии, которые проявлялись ограниченной дисплазией кортикальной пластинки в виде ее утолщения, стушеванности границы между серым и белым веществом, изменения рисунка прилегающих борозд и формы пораженной извилины [1, 9].
Анализируя данные литературы о туберозном склерозе, следует отметить следующие осложнения:
— эпилептические припадки;
— повышение внутричерепного давления и гидроцефалия;
— умственная отсталость;
— агрессивность или другие расстройства поведения;
— почечная недостаточность или геморрагический шок в связи с кровотечением из ангиомиолипом;
— пневмоторакс или обструктивные заболевания легких;
— сердечная аритмия или сердечная недостаточность;
— смерть.
Среди недавней важной информации о данной патологии следует выделить публикации об использовании рапамицина в ее лечении. Рапамицин (sirolimus) был впервые обнаружен как продукт деятельности бактерии Streptomyces hygroscopicus в образце почвы с острова Пасхи [25, 39]. Были установлены его иммуносупрессивные и антипролиферативные свойства, а в недавних исследованиях отмечались уменьшение частоты эпилептических припадков и регрессия астроцитом у больных с туберозным склерозом на фоне приема рапамицина [10, 12, 18, 23]. В настоящее время в США проводятся многочисленные клинические испытания с использованием рапамицина в лечении туберозного склероза у детей и взрослых [37].
Таким образом, вышеприведенные данные свидетельствуют о широким спектре изменений при туберозном склерозе и подтверждают мнение о природе заболевания как о полисистемной многоуровневой дизнейроонтогенетической патологии, что указывает на необходимость ее пренатальной диагностики (включая генетическое консультирование) и адекватного лечения.
Для иллюстрации значения своевременного определения патологии приводим историю болезни ребенка М., 2 года, находившегося на лечении в отделении детской психоневрологии Института педиатрии, акушерства и гинекологии АМН Украины.
Ребенок поступил в клинику с жалобами на эпилептические припадки, беспокойный сон, двигательную расторможенность в периоды бодрствования.
Припадки имели характер очаговых тонических судорог жевательных мышц, левых конечностей с поворотом головы влево, иногда сопровождающиеся наклоном вперед и падением, частотой до 20 раз в сутки в дневное и ночное время. Длительность припадков составляла от нескольких секунд до нескольких минут.
Анамнез жизни и заболевания: ребенок от второй беременности (1-я беременность прервана по медицинским показаниям — артериальная гипертензия на ранних сроках). На 34-й неделе беременности в отделении функциональной диагностики ГУ «ИПАГ АМН Украины» с помощью ультразвукового обследования диагностировано объемное образование полости сердца (с большой долей вероятности, рабдомиома). Было рекомендовано родоразрешение путем операции кесарева сечения в акушерской клинике института. Роды состоялись на 39-й неделе беременности. Родился доношенный мальчик массой 3130 г, длиной 52 см, с оценкой по шкале Апгар 8 баллов.
На 2-е сутки жизни в Научно-практическом центре детской кардиологии и кардиохирургии МЗ Украины проведено эхокардиографическое обследование, где выявлены множественные рабдомиомы левого желудочка, открытый артериальный проток, открытое овальное окно.
Учитывая, что кардиальные опухоли являются косвенным признаком туберозного склероза, для уточнения диагноза в возрасте 1 месяц проведено МРТ головного мозга, при котором субкортикально в белом веществе левой лобной доли и в кортикальных отделах на уровне семиовальных центров выявлены участки измененного МР-сигнала с неровными контурами, гиперинтенсивные на Т1-ВИ и практически изоинтенсивные на Т2-ТIRM-ВИ, размером до 1,2 см в диаметре, и округлые очаги субэпендимально в правом боковом желудочке (рис. 1 и 2). Также было проведено МРТ сердца для более четкой структурной идентификации выявленных при УЗИ изменений (рис. 3).
В 3 месяца появились эпилептические припадки в виде серий пропульсивных спазмов (до 7–8 серий в сутки).
По месту жительства ребенку последовательно назначались различные антиконвульсивные препараты. В связи с отсутствием эффекта от лечения был назначен вигабатрин (сабрил) (500 мг), являющийся эффективным антиконвульсивным препаратом для лечения туберозного склероза. Данный препарат принимается постоянно и в настоящий момент.
При поступлении в клинику состояние относительно удовлетворительное, повышена двигательная активность, психическое развитие задержанное, речь на уровне отдельных звуков. На коже туловища и конечностей отмечено 4 гипопигментированных пятна (первое пятно появилось в 6 месяцев) размерами 1 × 1,5 см и 1 × 1 см, одно пятно цвета «кофе с молоком» на туловище ниже пупка размером 2 × 1,5 см, участки «шагреневой кожи» под коленями. В неврологическом статусе очаговой патологии не выявлено.
Обследован: общий анализ крови — гемоглобин 77 г/л, гипохромия эритроцитов, анизо- и пойкилоцитоз, остальные показатели без патологии; биохимическое исследование крови без патологических изменений.
Ультразвуковое обследование органов брюшной полости не выявило очаговой патологии и объемных образований.
На глазном дне — умеренное сужение сосудов.
ЭЭГ (электроэнцефалография) показана на рис. 4 (до начала лечения) и рис. 5 (на фоне лечения).
Для контроля состояния церебральных структур было проведено МРТ головного мозга. Заключение: МРТ-картина характерна для туберозного склероза. Без существенной динамики по сравнению с предыдущим обследованием.
Данный случай демонстрирует классический пример течения туберозного склероза с его типичными начальными симптомами и подчеркивает необходимость своевременной диагностики данной патологии, начиная с момента определения рабдомиомы в сердце, с целью выбора и оптимизации лечебной тактики. Сочетание антиконвульсивной и нейрометаболической терапии позволило значительно уменьшить количество эпилептических припадков и улучшить развитие ребенка. Ребенок самостоятельно ходит и выполняет инструкции. В настоящее время единичные эпилептические припадки отмечаются в ночное время. Проводятся регулярное наблюдение за ребенком в динамике и коррекция терапии.
В описанном случае, возможно, именно ранняя диагностика, и, как следствие, продуманная врачебная тактика родоразрешения, и оптимальные лечебные назначения с применением антиконвульсивного препарата первого выбора — вигабатрина в сочетании с нейрометаболической терапией позволили достичь контроля над припадками и улучшить качество жизни ребенка.
В заключение следует еще раз отметить, что в представленном материале акцент сфокусирован на пренатальную диагностику. В отечественных публикациях и литературе стран ближнего зарубежья мы не встретили работ по пренатальной диагностике туберозного склероза. Решение этой важнейшей проблемы перинатальной неврологии вплотную связано как с научными, лечебными, так и с социально-этическими вопросами. Поскольку ультразвуковой метод имеет ограничения в определении мозговых структур плода, представляется необходимым при выявлении косвенных признаков туберозного склероза целенаправленное МРТ-исследование головного мозга плода.
- развитие шокового состояния
- падение артериального давления
- вегетативные расстройства (обильный пот, озноб, сердцебиение)
Высокое артериальное давление, головные боли, утомляемость, кожный зуд.
Поверхностное дыхание, бледность кожных покровов рецидивирующий пневмоторакс. Симптомы могут свидетельствовать о развитии патологического процесса в легких (лимфангиомиоматоз легких).
Продолжение.