А.А. Устинович, Т.М. Галица, Е.Е. Жеребилова, С.В. Кузьмина, Г.В. Леус, А.Н. Ракуть
СИНДРОМ КЛИППЕЛЯ-ФЕЙЛЯ
Белорусский государственный медицинский университет,
УЗ 3-я городская детская клиническая больница
A.A. Ustinovich, T.M. Galitsa, E.E. Jerebilova, S.V. Kuzmina, G.V. Leus, A.N. Racut.
KLIPPEL - FEIL SYNDROME
В 1912 году французские врачи М. Klippel (невропатолог) и Andre Feil (рентгенолог) описали врожденный порок развития позвоночника, характеризующийся деформацией (укорочением) шеи, обусловленной уменьшением числа шейных позвонков, их сращением или меньшими размерами.
Различают три типа деформации: первый тип - уменьшение общего числа шейных позвонков; второй тип - синостоз всего спаянного в единую кость шейного отдела позвоночника с затылочной костью и верхними грудными позвонками; сочетание 1 или 2 типа с синостозом нижнегрудных и поясничных позвонков. Часто деформация сочетается с незаращением дужек позвонков (spina bifida cervicalis), наличием шейных ребер, синхондрозом лопаток с позвоночником при высоком их стоянии (болезнью Шпренгеля).
В большинстве случае синдром спорадичен, имеются данные о его генетической гетерогенности, например, 2 тип наследуется аутосомно-доминантно, а 3 тип - аутосомно-рецессивно.
У больных отмечается укорочение и ограничение подвижности шеи, низкая граница роста волос на затылке, кифосколиоз. Укорочение шеи придает пациентам особый вид - «человека-лягушки». В тяжелых случаях подбородок упирается в грудину, мочки ушей касаются плеч, затрудняется дыхание и глотание. У части больных могут быть крыловидные складки шеи, пороки развития мышц плечевого пояса. Лопатки широко разведены, часто укорочены. В большинстве случаев деформация безболезненна, но иногда сопровождается синдромом сдавления шейных корешков спинного мозга. Возможны асимметрия лица, аномалии зубов, микроцефалия, гидроцефалия, спинно-мозговая грыжа, пороки ребер, лучевой кости и ее производных, постаксиальная полидактилия. В 45% случаев наблюдаются гипоплазия и дистопия почек, в 25% - глухота, в 17 - 20% - расщелина неба, в 15% - пороки сердца. Также характерны пороки развития нервной системы и умственная отсталость. Со стороны глаз наблюдаются паралитическое косоглазие, гиперметропия, нистагм, синдром Горнера и Щтиллинга-Тюрка-Дуана. Также характерны: слабость рук и ног, переходящая позднее в спастические и паралитические параплегии и тетраплегии, нарушения функций симпатического отдела нервной системы, зеркальные движения конечностей (из-за возможного отсутствия перекреста пирамид), глухота, эпилептические припадки, приступы головной боли.
Диагностика синдрома основана на триаде клинических симптомов: укорочение шеи, наблюдаемое с рождения, низкая граница роста волос на шее и ограничение подвижности головы. Для уточнения типа деформации проводят рентгенологическое исследование шейного и грудного отделов позвоночника в прямой и боковой проекциях. На рентгенограммах чаще выявляют сращение 4-6 шейных позвонков в сплошную малодифференцированную костную массу. Иногда тела позвонков сливаются лишь частично и тогда можно проследить узкие полоски просветления - недоразвитые межпозвоночные диски. При полном синостозе блокированными оказываются тела, дужки и отростки позвонков. Частичный синостоз вызывает в процессе роста искривление позвоночника в сагиттальной или фронтальной плоскости.
Дифференциальный диагноз проводят с туберкулезным спондилитом верхних шейных позвонков, двусторонней и односторонней формами мышечной кривошеи (особенно при отсутствии эффекта от консервативного лечения).
Лечение, как правило, консервативное (ЛФК, массаж), направлено на улучшение осанки и предупреждение вторичных деформаций. При возникновении компрессии корешков спинного мозга, например шейными ребрами, их резецируют. Для устранения болевого синдрома назначают мягкий воротник типа Шанца, анальгезирующие средства, физиотерапию.
Витальный прогноз благоприятный при отсутствии пороков внутренних органов, однако у больных возникают серьезные косметические и функциональные проблемы.
В приводимом клиническом наблюдении выявлено редкое сочетание синдрома Клиппеля - Фейля и тератомы спинного мозга.
Девочка П. родилась от 1 беременности, 1 родов, в сроке 40 недель. Беременность отягощена хронической фето-плацентарной недостаточностью, в родах - раннее излитие околоплодных вод., угрожающий разрыв промежности, безводный период . 2 ч. 50мин. Масса тела при рождении - 3900, длина - 55 см, оценка по шкале Апгар 8/9 баллов. Выписана домой на 3 сутки жизни. На следующий день (4 сутки жизни) на врачебном патронаже отмечены жалобы на снижение аппетита, вялость, осиплость голоса, отсутствие движений в правой ручке и с диагнозом «ОРИ, перелом ключицы?» направлена в стационар.
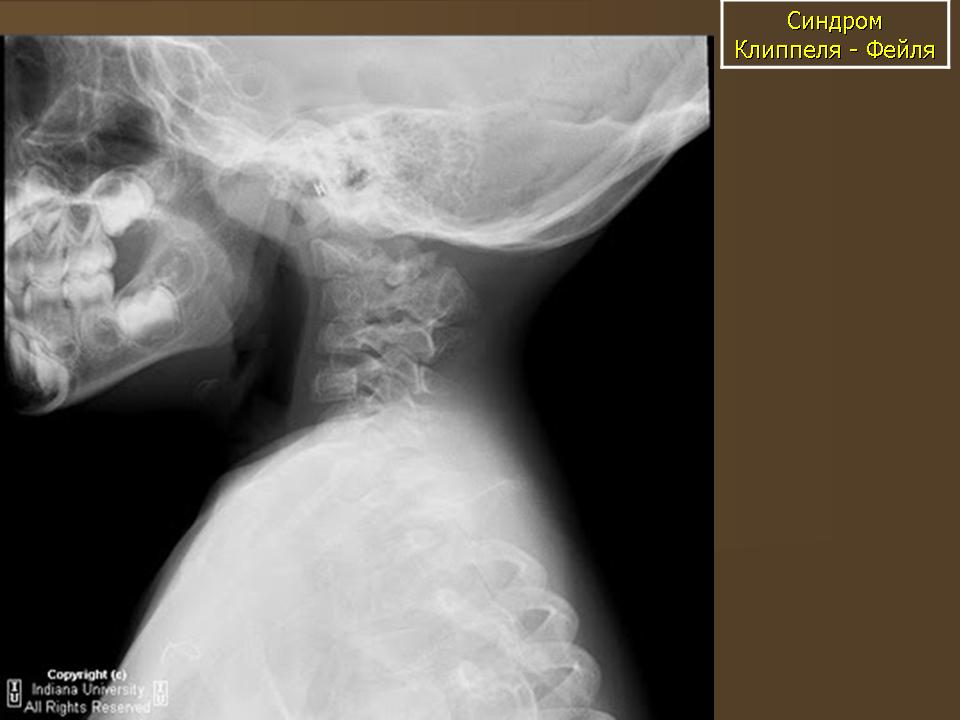
С целью уточнения характера патологии ребенку выполнены обзорная рентгенограмма скелета, (рис.1), КТ и МРТ ЦНС.

Заключение КТ головного мозга: выявляется отек белого вещества головного мозга, умеренно выраженная внутренняя гидроцефалия Гипотрофия червя мозжечка. Кистоподобный тяж в проекции продолговатого мозга.
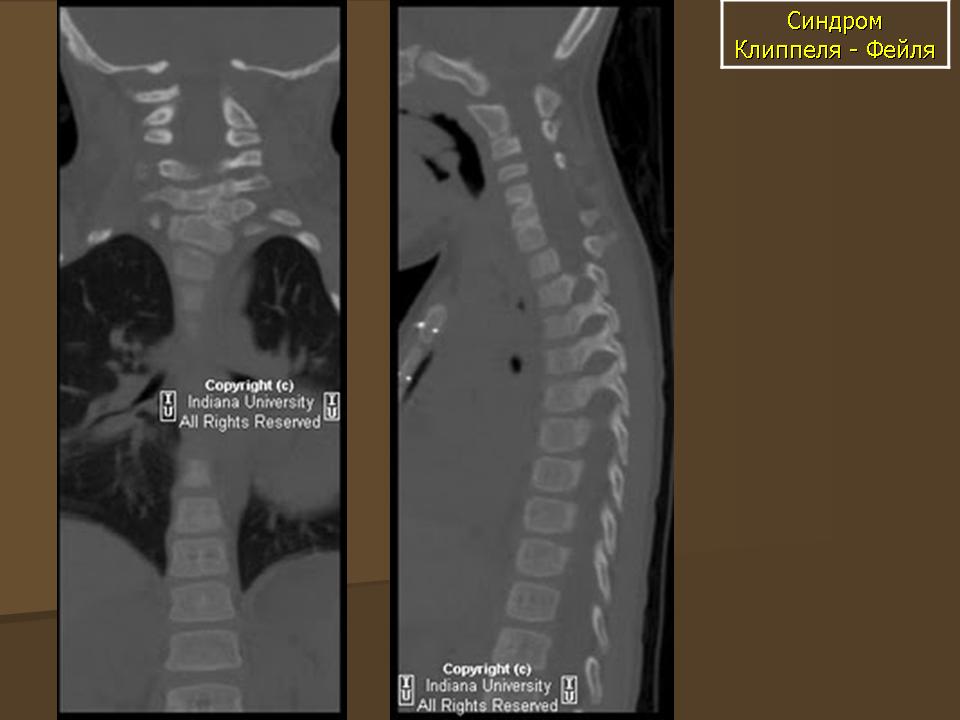
КТ шейного и грудного отдела позвоночника: врожденный костный блок тел 2.3.7. грудных позвонков с передней расщелиной 3 и 4 грудных позвонков. Поперечные отростки С5, С6, С7 позвонков слиты к костные конгломераты с обеих сторон. Аномалия сращения 3, 4 и 5 ребра с обеих сторон. Правая ключица вогнута в средних отделах. Широкая расщелина задней стенки позвоночного канала на протяжении С6 - Тh4, максимальной шириной до 21мм в коронарной плоскости. В проекции позвоночного канала на участке С5 -Тh2 определяется интрадуральное изогиперинтенсивное объемное образование 20×15мм, вызывающее деформацию (вогнутость) тел прилежащих позвонков: Заключение: КТ признаки объемного патологического образования позвоночного канала на участке С5 - Тh2 позвонков. Врожденные костные аномалии.
МРТ шейного отдела позвоночника: интрамедулярно на уровне С5 - Тh2 определяется объемное образование 20×15мм с бугристыми контурами, интенсивно накапливающее контрастное вещество, Имеет место врожденная аномалия спинного мозга. С уровня продолговатого мозга он раздвоен, истончен, диаметр 1,5×2 мм до уровня вышеописанного патологического образования. Гипоплазированы обе гемисферы мозжечка, его червь. В грудном отделе спинного мозга на уровне Тh7, Тh8, Тн12 -L1 определяется гипоинтенсивный сигнал (нельзя исключить сирингомиелию). Заключение: МРТ проявления интрамедуллярного объемного образования на уровне С5 -Тh2 спинного мозга. Врожденная аномалия спинного мозга (его раздвоенн6ость в шейном отделе) Сирингомиелия в грудном отделе, гипоплазия червя мозжечка, его гемисфер.
На 8 сутки жизни ребенок был переведен в нейрохирургическое отделение 9 городской клинической больницы с диагнозом: Объемное образование спинного мозга (зрелая тератома) на уровне С7 -Тн3 позвонков. ВПР ЦНС: болезнь Клиппеля - Фейля с гипоплазией мозжечка. Тетрапарез, больше справа. Бульбарный синдром.
На 11 сутки жизни ребенку была проведена ляминэктомия С7 - Тh3, тотальное удаление опухоли.
Гистологическое заключение: зрелая тератома.
В послеоперационный период в неврологическом статусе сохранялись бульбарные нарушения в виде нарушения глотания, тетрапарез, глубже справа, сохранялся длительный субфебрилитет.
Получала медикаментозное лечение: аналгин, цефатоксим, амикацин, аугментин, биофлор, дексаметазон, преднизолон, этамзилат, диалакт, панкреатин, инфузии СЗП, эритроцитной массы.
В дальнейшем, на 3-м месяце жизни у ребенка развился эпилептический синдром с наличием генерализованных припадков. На фоне респираторных проблем, обусловленных деформацией грудной клетки (рис. 2) и неврологической симптоматикой, сформировались кардиомегалия и легочная гипертензия II степени, НКI степени. В дальнейшем ребенок страдал глубокой задержкой психомоторного развития, также у него сохранялся судорожный синдром.
Таким образом, в данной клинической ситуации наблюдается редкое сочетание синдрома Клиппеля - Фейля с тератомой шейно-грудного отдела позвоночника, что обусловило развитие у ребенка тяжелых неврологических и соматических нарушений.
Что такое синдром короткой шеи (синдром Клиппеля-Фейля)
Синдром короткой шеи (см. фото) представляет собой врожденный сколиоз шейного и верхней части грудного отдела позвоночника, который возникает вследствие слияния позвоночных сегментов, расщепления и промежуточного включения рудиментов позвонков.
Помимо кривизны шея укорочена, а подвижность ее ограничена (короткая шея). Граница роста волос на затылке находится низко, а в выраженных случаях отсутствует четкое разграничение между головой и туловищем (рис.2). В этом случае говорят о синдроме Клиппеля-Фейля.
Указаниями на заболевания соединительной ткани, костей и суставов часто являются необычные, значимые для "моментального" диагноза изменения кожи, глаз и осанки пациента.
При синдроме Клиппеля-Фейля, деформации шейного отдела позвоночника из-за сросшихся шейных позвонков, почти постоянно наблюдается крыловидная складка (птеригий, рис.1), которая присутствует также и при синдроме Ульриха-Тернера.
Факультативными аномалиями при этом синдроме являются аплазия грудино-ключично-сосцевидной мышцы, волчья пасть, синдактилия, врожденное слабоумие и врожденные пороки сердца (10- 20 % случаев).
Синдром Клиппеля-Фейля является особой формой синдрома короткой шеи.
Ф.B.Тишeндop
Синдром Клиппеля-Файля
Синдром Клиппеля-Файля (другое название - синдром короткой шеи) заключается в конкресценции (сращении шейных позвонков). Иногда между собой сращиваются не только шейные, но и верхние грудные позвонки.
Эта аномалия развития позвоночника проявляется выраженным укорочением шеи, снижением границы роста волос, ограничением движений при поворотах головы в сторону, «гордой посадкой головы» (голова слегка отклоняется кзади). В некоторых случаях у пациентов с синдромом Клиппеля-Фейля имеются выраженные кожные складки от ушей до плеч.
Синдром короткой шеи нередко сочетается с другими аномалиями развития позвоночника (шейные ребра, Spina bifida), сердечно-сосудистой и нервной системы. Неврологические симптомы могут отсутствовать. В некоторых случаях возможно сдавление корешков, сопровождающееся нарушением чувствительности, снижением силы рук или парезами.
Синдром Клиппеля-Фейля (синдром короткой шеи) представляет собой врожденные аномалии и сращение шейных позвонков, часто сочетается с синдромом Ольеника. Возможна неполная дифференцировка шейных позвонков и уменьшение их числа, иногда количество их не превышает четырех. В клинической картине характерна триада: короткая шея («человек без шеи», «шея лягушки»), низкая граница роста волос на шее, значительное ограничение подвижности головы. В тяжелых случаях подбородок упирается в грудину, мочки ушных раковин касаются надплечий, иногда - кожные складки идут от ушных раковин к плечам. Может сочетаться с гидроцефалией, элементами бульбарного синдрома, вертебрально-базилярной сосудистой недостаточностью, проводниковыми нарушениями, высоким стоянием лопаток, проявлениями дизрафического статуса. По данным рентгенологических исследований, выделяют две крайние формы синдрома Клиппеля-Фейля: 1) атлант слит с другими шейными позвонками, общее количество которых в связи с этим уменьшено, обычно их не более 4; 2) признаки синдрома Ольеника и синостоз шейных позвонков, высота их тел снижена. Нередко сочетается с платибазией, возможны другие пороки развития. Описали синдром в 1912 г. французские невропатологи М. Klippel (1858-1942) и A. Feil (род. в 1884 г.).
СИНДРОМ КЛИППЕЛЯ-ФЕЙЛЯ
Врожденное сращение двух или более шейных позвонков. Различают сращение только тел позвонков (врожденный блок позвонков) и сращение всех элементов позвонков (включая задние структуры). Возникает в результате нарушения нормальной сегментации шейных сомитов на 3-8 нед после зачатия. Вовлеченные тела позвонков часто уплощены и дисковые промежутки между ними гипоплазированы или отсутствуют. Могут наблюдаться половинные позвонки. Корешковые отверстия меньше, чем в норме и имеют овальную форму. Шейный стеноз наблюдается редко. Полное отсутствие задних элементов с увеличением БЗО и фиксированным положением переразгибания шеи называется иниэнцефалией и встречается редко. Частота синдрома Клиппеля-Фейля неизвестна ввиду его редкости, а также того, что часто он протекает бессимптомно.
Классическая клиническая триада (все элементы встречаются в <50% случаев): низкая задняя линия роста волос, укороченная шея (бревиколлис), ограничение подвижности шеи (может быть незаметным, если имеется сращение <3 позвонков, если сращение ограничено только нижними шейными позвонками или если имеется компенсаторная гиперподвижность нефиксированных сегментов). Ограничения подвижности более заметны при вращении, чем при передне-заднем сгибании/разгибании или боковом сгибании.
Синдром может наблюдаться в сочетании с другими врожденными аномалиями шейного отдела, т.к. базиллярная импрессия и атланто-затылочное сращение. Кроме того, могут наблюдаться сопутствующий сколиоз (60% случаев), асимметрия лица, тортиколлис, сморщивание кожи шеи (в тяжелом случае называется pterygium шея), деформация Спренгеля в 25-35% случаев (высокое расположение лопатки в результате нарушения ее опущения из места образования высоко на шее до нормального положения примерно в то время, когда возникает синдром Клиппеля-Фейля), синкенизии (зеркальные движения, преимущественно в кистях, но иногда и в целой руке) и реже параличи лицевой мускулатуры, птоз, расщепленное или высокое небо. Возможны также системные врожденные аномалии: моче-половой системы (наиболее частой является одностороннее отсутствие почки), сердечно-сосудистые, ЦНС и в ∼30% случаев – глухота (в связи с дефектами развития косточек внутреннего уха).
Не удалось выявить симптомы, непосредственно связанные со сращением позвонков, однако, несращенные сегменты, которые могут быть гипермобильными и приводить к нестабильности или дегенеративным артритным изменениям, могут вызывать симптомы (которые наблюдаются реже при коротких сращениях).
Лечение
Обычно направлено на определение и лечение сопутствующих системных аномалий. Необходимо провести обследование ССС (ЭКГ), РГК, УЗИ почек. Для наблюдения за нестабильностью показаны серийные спондилограммы шейного отдела со сгибанием/разгибанием в боковой проекции. Иногда, несмотря на возможный риск нарушения в дальнейшем подвижности шеи, разумно осуществить фиксацию нестабильных несращенных сегментов.
Гринберг. Нейрохирургия
Klippel-Feil Syndrome
http://www.ajnr.org/content/29/2/306.full
Klippel-Feil Syndrome
http://radsource.us/klippel-feil-syndrome/
Рисунок 1