Врожденные пороки сердца у детей с генными синдромами.
Г.Э. СУХАРЕВА, Крымский государственный медицинский университет им. С.И. Георгиевского, кафедра педиатрии с курсом детских инфекционных болезней, Республиканская детская клиническая больница, г. Симферополь
Резюме
Проведено наблюдение за 56 детьми в возрасте 0-18 лет с врожденными пороками сердца и ассоциированной наследственной патологией, у 20 из них отмечались генные синдромы. Выявлены особенности течения врожденных пороков сердца и сосудов у больных с наследственными заболеваниями. В представленной работе впервые в отечественной литературе описано поражение сердца у двух детей с синдромом Секкеля. Проведенные исследования доказывают необходимость осмотра детей с врожденными пороками сердца врачом-генетиком и обязательного обследования больных с синдромальной патологией для исключения врожденных аномалий развития сердечно-сосудистой системы.
Наследственность - омнибус, в котором нас сопровождают наши предки; то и дело кто-нибудь из них высовывается оттуда, ошеломляя нас своим появлением.
Оливер Холмс
В Украине врожденные сердечно-сосудистые заболевания у детей представляют собой все более серьезную проблему здравоохранения. С начала 90-х годов частота заболеваний органов кровообращения в детском возрасте возросла в 2,5-3 раза [8]. Одной из задач клинициста является поиск этиологического фактора сердечной патологии. За последние годы произошел существенный пересмотр взглядов на природу патологии сердца. Если до недавнего времени в качестве ведущей причины рассматривалось влияние вирусов на кардиогенез с последующим формированием врожденных пороков сердца (ВПС) или развитием воспаления, то научные исследования последних лет свидетельствуют об огромной роли в формировании различной патологии сердца генетических факторов [1, 3, 4, 6, 9, 13, 15, 20]. Врожденные пороки сердца и сосудов представляют с генетической точки зрения весьма гетерогенную группу, встречаясь либо в изолированном виде, либо входя в состав множественных врожденных пороков развития (МВПР), а также моногенных (аутосомно-доминантных или аутосомно-рецессивных) или хромосомных синдромов [2, 7, 11, 12, 14, 16, 17, 19, 23].
Заболевания сердечно-сосудистой системы с учетом влияния генетических факторов Н.А. Белоконь [5] подразделила на следующие группы:
1. Сердечно-сосудистая патология при наследственных синдромах.
2. Сердечно-сосудистая патология при хромосомных аномалиях.
3. Семейные заболевания, при которых сердце является единственной (основной) мишенью поражения.
4. Болезни с наследственным предрасположением, при которых наряду с генетическими большое значение имеют факторы влияния среды.
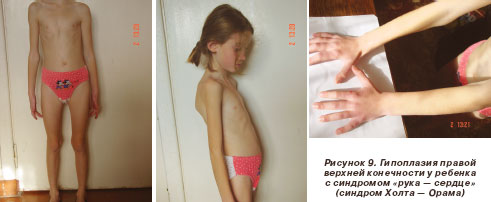
Сопутствующая патология может иметь различное влияние на состояние ребенка, часто определяя прогноз более существенно, чем сам ВПС: различные наследственные заболевания могут значительно осложнять развитие ребенка, затруднять хирургическое лечение порока сердца и приводить к осложнениям в пред- и послеоперационном периоде, а в некоторых случаях даже могут ставить под сомнение целесообразность хирургической коррекции. В частности, кардиохирург должен знать, что при синдроме Холта -- Орама [22] затруднена пункция периферических артерий в связи с патологией лучевой кости и, соответственно, аномальным расположением сосудов. Послеоперационный период синдромов Беквита - Видемана и Ди Джорджи может сопровождаться судорогами гипогликемической и гипокальциемической природы, а также проблемами инфекционного характера и т.д.
Изучение сердечно-сосудистых заболеваний у детей с ассоциированной наследственной патологией является актуальным, так как своевременная диагностика синдромальной патологии, знание этиологии и патогенеза развивающихся специфических осложнений, их профилактика и лечение могут улучшить прогноз и качество жизни пациентов с ВПС.
Цель исследования: выявить особенности течения врожденных пороков сердца и сосудов у больных с генными синдромами.
Нами проведено наблюдение за 56 детьми с ВПС в возрасте 0-18 лет с заболеваниями сердечно-сосудистой системы и ассоциированной наследственной патологией (у 20 из них отмечались генные синдромы), прошедшими стационарное лечение в отделениях реанимации новорожденных, патологии новорожденных, врожденной и наследственной патологии, кардиоревматологии РДКБ и находящимися на диспансерном учете в Крымском республиканском медико-генетическом центре за период 2001-2007 гг.
Методы исследования: клинические; общие лабораторные методы; ЭКГ в покое и при физической нагрузке в 12 стандартных отведениях; холтеровское мониторирование ЭКГ; рентгенография органов грудной клетки в прямой и боковой проекциях; эхокардиография сердца и магистральных сосудов; пульсоксиметрия; кариотипирование; генеалогический метод; генетические биохимические методы.
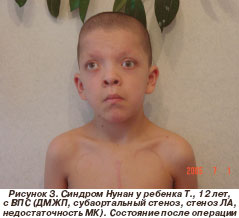
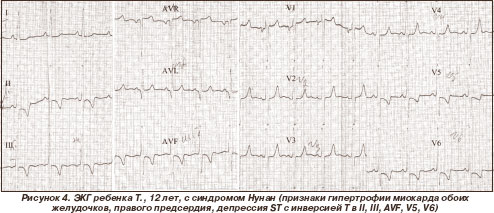
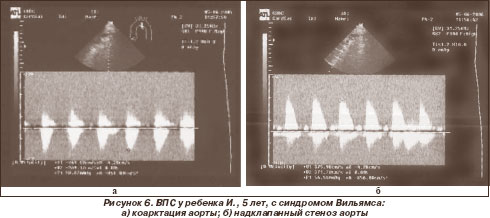
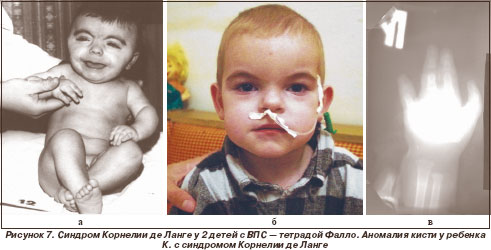
Среди генетических синдромов, ассоциированных с поражением сердца, преобладали синдром Марфана - у 4 больных (20 %) (рис. 1, 2), синдромы Нунан (рис. 3, 4), Вильямса (рис. 5, 6), Ивемарка (рис. 16) - по 3 случая (по 15 %), синдромы Секкеля и Корнелии де Ланге (рис. 7) - по 2 случая (по 10 %). В единичных случаях (по 5 %) выявлены синдромы Смита - Лемли - Опитца (рис. 8), Холта - Орама (рис. 9), Крузона.
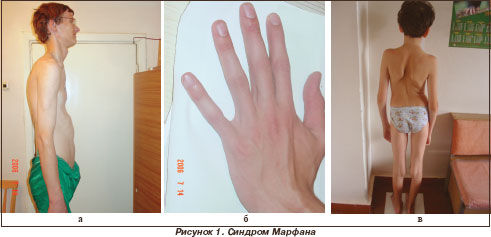



У всех больных с синдромом Марфана имеется расширение восходящей аорты как признак начинающейся аневризмы аорты. Несмотря на то что в настоящее время нет показаний к хирургическому лечению, дети требуют постоянного контроля кардиолога для проведения своевременного оперативного лечения.
Все дети с синдромом Вильямса были прооперированы: устранены КА, надклапанный стеноз аорты. Однако у 1 ребенка во время операции была диагностирована гипоплазия аорты на большом протяжении, что является противопоказанием для ее устранения. Дети находятся под наблюдением кардиолога.
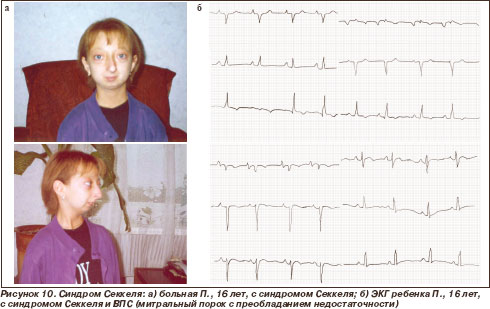
Мы наблюдали 2 детей с редким синдромом Секкеля, описанным в единичных источниках в зарубежной литературе [21, 24, 25]. Так, M.R. MacDonald et al. описали всего 60 наблюдений данного синдрома на протяжении 40 лет, а поражение сердца при синдроме Секкеля в зарубежной литературе описано лишь в нескольких работах. В наших наблюдениях у больных с синдромом Секкеля была диагностирована прогрессирующая митральная недостаточность, у 1 ребенка - в сочетании с митральным стенозом (рис. 10). Больная умерла в возрасте 18 лет, так как родители отказались от хирургического лечения.
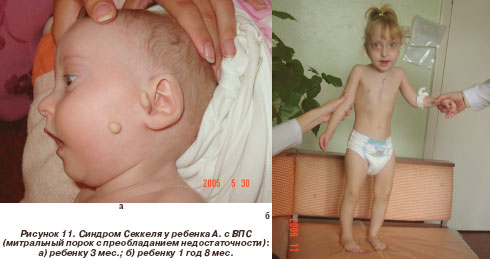
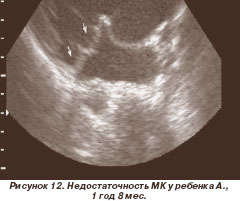
У второго ребенка (рис. 11а) в возрасте 3 месяцев диагностирована недостаточность МК (+). При наблюдении в динамике зафиксировано прогрессирование данных изменений до МК (++) (рис. 12) в возрасте 1 год 8 мес. (рис. 11б). Ребенку будет показано оперативное лечение при дальнейшем прогрессировании митральной недостаточности.
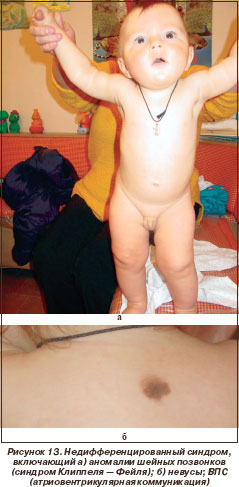
В 2 случаях недифференцированных МВПР проводился синдромальный поиск. В первом случае возникла необходимость синдромального поиска у ребенка 2 лет с ВПС (атриовентрикулярная коммуникация).
У ребенка отмечаются аномалия развития шейных и верхнегрудных позвонков («человек без шеи»), задержка роста (синдром Клиппеля - Фейля). Больной был направлен на хирургическое лечение ВПС (рис. 13).
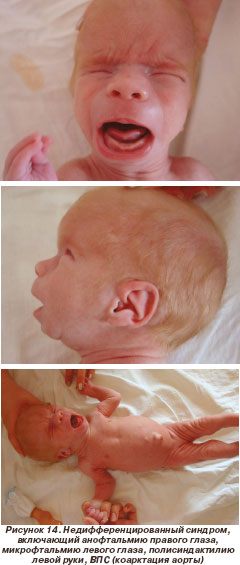
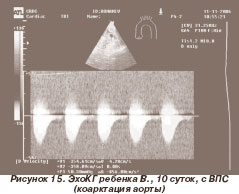
Во втором случае (рис. 14) ребенку с недифференцированным синдромом в возрасте 6 мес. по жизненным показаниям была проведена хирургическая коррекция ВПС (устранена КА). Результат операции удовлетворительный. В настоящее время ребенок находится под наблюдением окулистов.
Синдром Ивемарка (рис. 16), или синдром гетеротаксии [10, 18], часто сочетается с единственным желудочком сердца (ЕЖС), что нашло свое отражение в классификации ВПС: SV, heterotaxia syndrome (синдром Ivemark's, изометризм, синдром асплении/полисплении).
Все дети с синдромом Ивемарка в наших наблюдениях умерли в неонатальном периоде в связи со сложностью хирургической коррекции ВПС на фоне генетического синдрома.
Таким образом, состояние детей с синдромальной патологией определяется не только тяжестью поражения сердца, различные наследственные заболевания могут приводить к дисфункции других органов и систем, тем самым затруднять терапевтическое и хирургическое лечение порока сердца. Проведенные исследования доказывают необходимость осмотра детей с ВПС врачом-генетиком и обязательного обследования больных с синдромальной патологией для исключения врожденных аномалий развития сердечно-сосудистой системы. Своевременная диагностика наследственного синдрома поможет родителям получить достоверную информацию о прогнозе жизни и развития ребенка, принять адекватное решение о возможности и особенностях его воспитания в семье, узнать генетический риск при повторном деторождении и принять меры дородовой профилактики, а врачам - определить тактику ведения больного, в том числе оценить возможность и, главное, целесообразность проведения кардиохирургической коррекции.
Экономисты и генетики подсчитали, что затраты на проведение своевременной диагностики и коррекции наследственных заболеваний будут в 25 раз меньшими, чем расходы на содержание и лечение людей с наследственной патологией, а также на пособия по инвалидности.
«Если детей с врожденным уродством показать неподготовленным людям, это может произвести страшное впечатление. Подобный прием я бы отнес к числу запрещенных. Но коль есть угроза, лучше показать. Пусть Он и Она знают, что ожидает их потомство», - заметил заведующий отделом Института молекулярной биологии и генетики Национальной академии наук, академик АМН, член-корреспондент НАН Украины, профессор В. Кордюм.
Врожденные пороки сердца
Дефект межжелудочковой перегородки (ДМЖП)
По частоте занимает первое место, составляя 10—25 % от всех ПСВ. Дефект располагается в мембранозной, реже в мышечной части перегородки.
Нарушения гемодинамики обусловлены сбросом крови из левого желудочка в правый, объем и направление которого зависят от величины дефекта и разницы давления в малом и большом круге кровообращения. Дефекты небольших размеров (болезнь Толочинова—Роже) сопровождаются незначительным сбросом, не вызывающим выраженных нарушений гемодинамики. Общее состояние больных удовлетворительное, дети развиваются нормально. Размеры сердца не увеличены или выявляется небольшое увеличение левого желудочка.
Единственным проявлением порока может быть грубый, скребущий систолический шум в третьем-четвертом межреберье слева. При больших дефектах со значительным сбросом крови наблюдается гиперволемия малого круга кровообращения, приводящая к легочной гипертензии. Последняя является основным фактором, определяющим тяжесть клинических проявлений порока, течение и прогноз.
У больных с большими дефектами перегородки клинические признаки порока выявляются в первые месяцы жизни. Отмечаются одышка, тахикардия, застойные хрипы в легких, увеличение печени, селезенки и др. Дети часто болеют пневмонией, развивается гипотрофия. Рано появляется сердечный горб, верхушечный толчок усилен. В третьем-четвертом межреберье слева определяется систолическое дрожание. Границы сердца увеличены влево. Выслушивается интенсивный систолический шум в третьем-четвертом межреберье слева, который хорошо проводится вправо от грудины. На верхушке может быть мезодиастолический шум как проявление относительного стеноза левого венозного устья; II тон над легочной артерией усилен и расщеплен. Прогрессирование легочной гипертензии приводит к увеличению пререгрузки правого желудочка, появлению признаков его гипертрофии, которая в тяжелых случаях может быть преобладающей. При очень высокой легочной гипертензии выравнивается давление в желудочках и возникает перекрестный сброс крови.
На ЭКГ при умеренной легочной гипертензии регистрируются признаки гипертрофии левого желудочка, при высокой — обоих желудочков или правого желудочка. Рентгенологическая картина также соответствует степени легочной гипертензии. При умеренной легочной гипертензии увеличен левый желудочек, выбухает дуга легочной артерии и переполнено артериальное русло легких. При значительной легочной гипертензии сердце увеличено за счет обоих желудочков, выражено выбухание легочной артерии, аорта гипоплазирована. Усиление легочного рисунка связано с артериальной гипертензией и венозным застоем.
Лечение. При ЗСН показаны сердечные гликозиды, периферические вазодилататоры, при необходимости назначают диуретики. Возможно спонтанное закрытие дефекта, которое чаще происходит в возрасте до 4 лет. Хирургическое лечение проводится при прогрессирующей легочной гипертензии и стойкой ЗСН.
Дефект межпредсердной перегородки (ДМПП)
Встречается у 8—16 % детей, родившихся с ПСВ.
Различают первичные ивторичные ДМПП.
Первичные дефекты расположены низко над атриовентрикулярными клапанами, вторичные — в области овального окна. Гемодинамические нарушения связаны со сбросом крови из левого предсердия в правое, величина которого зависит от размера дефекта и разницы давления в предсердиях. При малых дефектах нарушения кровообращения не возникает и порок выявляется поздно. У больных с большими дефектами сброс крови приводит к перегрузке правых отделов сердца, увеличению кровотока в легких и развитию легочной гипертензии. При ДМПП легочная гипертензия развивается относительно редко, особенно «склеротическая» ее форма.
Больные с ДМПП чаще жалоб не предъявляют, в физическом развитии не отстают, ЗСН возникает редко. Границы сердца увеличены вправо, во втором межреберье слева выслушивается систолический шум, чаще неинтенсивный, с ограниченной зоной распространения; II тон над легочной артерией расщеплен, усилен за счет легочного компонента.
На ЭКГ определяются признаки гипертрофии правого желудочка (или блокада правой ножки пучка Гиса, чаще неполная), реже и правого предсердия.
На рентгенограммах выявляются увеличение правых отделов сердца, выбухание дуги легочной артерии, переполнение артериального русла легких.
Лечение. Хирургическое; операция показана при легочной гипертензии и наличие признаков ЗСН.
Коарктация аорты
Сужение аорты или ее облитерация на ограниченном участке в области дуги, грудного или брюшного отдела. Сужение чаще локализуется в области перехода аорты в нисходящую часть (в «типичном» месте). Различают «детский» и «взрослый» типы коарктации аорты. Первый отличается от второго наличием открытого аортального порока и сужением аорты на большом участке ее дуги.
Нарушения гемодинамики зависят от уровня, степени сужения аорта и развития коллатералей, а также типа коарктации. Наиболее характерны жалобы на одышку, головные боли, головокружения, боли в икроножных мышцах при ходьбе. К ценным диагностическим признакам относится разница наполнения пульса и величины систолического давления на верхних и нижних конечностях. На руках отмечаются высокое наполнение пульса и повышение систолического давления, доходящее в ряде случаев до 200 мм рт. ст. и выше. На ногах пульс слабый, артериальное давление понижено, иногда не определяется.
В ряде случаев при коарктации аорты артериальное давление на руках в пределах нормы. Физическое развитие больных часто не нарушено, но обращает внимание диспропорция тела: при хорошо развитом плечевом поясе нижняя половина тела относительно недоразвита.
Границы сердца умеренно расширены влево. При аускультации отмечается систолический шум во втором-третьем межреберье слева. Шум более выражен в межлопаточном пространстве слева от позвоночника.
На ЭКГ выявляется гипертрофия левого желудочка, у детей раннего возраста чаще правого желудочка.
Рентгенологически отмечаются увеличение левого желудочка, постстенотическое расширение аорты, у детей старшего возраста — узурация нижних краев ребер.
Лечение. Хирургическое, оптимальным сроком для операции считается возраст 7—15 лет. В более раннем возрасте хирургическая коррекция порока проводится при наличии симптомов ЗСН.
Открытый артериальный проток (ОАП)
Во внутриутробной жизни обеспечивает кровоснабжение плода. Он закрывается в первые дни или месяцы после рождения ребенка. Функционирование протока более 2 нед внеутробной жизни расценивается как ПСВ, частота которого составляет 10—30 %.
Клиническая картина. Имеет значение состояние гемодинамики, нарушение которого связано со сбросом крови через проток из аорты в легочную артерию. Величина сброса зависит от размера протока, угла его отхождения от аорты и разницы сопротивления в малом и большом круге кровообращения. Возникает переполнение малого круга кровообращения. Левый желудочек выполняет повышенную работу, развивается его гипертрофия. При развитии высокой легочной гипертензии в результате перегрузки возникают дилатация и гипертрофия правого желудочка.
При отсутствии выраженных нарушений гемодинамики течение ОАП чаще бессимптомное. У больных со значительным сбросом крови рано развивается легочная гипертензия, в связи с чем они часто болеют пневмониями, отстают в физическом развитии, при этом прогрессируют симптомы ЗСН. Границы сердца при ОАП расширены влево, при высокой легочной гипертензии — и вправо. Во втором межреберье слева выслушивается непрерывный систолодиастолический шум, обусловленный движением крови в период систолы и диастолы в одном направлении. В первые месяцы жизни ребенка и при значительной легочной гипертензии сброс крови через проток происходит только во время систолы, поэтому выявляется лишь систолический шум; II тон над легочной артерией усилен и расщеплен.
На ЭКГ определяются признаки гипертрофии левого желудочка, при легочной гипертензии — обоих желудочков.
Рентгенологически выявляются увеличение левого желудочка, выбухание дуги легочной артерии, усиление пульсации аорты и легочной артерии, переполнение малого круга за счет артериального русла. При высокой легочной гипертензии отмечается увеличение и правого желудочка.
Лечение. Хирургическое, операция показана при легочной гипертензии и наличии признаков ЗСН. Возможно спонтанное закрытие протока в первые месяцы жизни, особенно у недоношенных детей, у которых для его закрытия в последние годы применяют индометацин (внутривенно в дозе 0,1 мг/кг 3—4 раза в день в первые 2 недели жизни). При ЗСН используют сердечные гликозиды и диуретики, проводят кислородотерапию, переливание крови.
Стеноз аорты
От всех ПСВ составляет 2—7 %.
Бывает клапанным, подклепанным и надклапанным. Первый встречается наиболее часто.
Степень стеноза обусловливает тяжесть нарушения гемодинамики и соответственно клинических проявлений порока. При резком стенозе симптомы порока развиваются в раннем детском возрасте: бледность кожных покровов, тахикардия, признаки ЗСН.
При небольшом и умеренном стенозе клинические проявления развиваются постепенно, они наиболее четко проявляются у детей школьного возраста. К ним относятся одышка, боли в области сердца, головокружения При резком стенозе возможны приступы потери сознания (синкопе). Границы сердца расширены влево, как правило, незначительно, часто они не увеличены. Определяется систолическое дрожание во втором межреберье справа и в яремной ямке. При аускультации в этой области выслушивается грубый, скребущий систолический шум, иррадиирующий на сосуды шеи.
На ФКГ он имеет ромбовидную форму, аортальный компонент II тона отсутствует или значительно снижен.
На ЭКГ — признаки гипертрофии левого желудочка.
На рентгенограммах определяются увеличение левого желудочка и расширение восходящей аорты.
Лечение. Хирургическое: проводится вальвулотомия, балонная дилатация клапанного стеноза или протезирование (у детей старшего возраста) аортального клапана.
Стеноз легочной артерии изолированный
Составляет 0,9—6,7 % от общего числа ПСВ. Встречается в виде разных анатомических вариантов, среди которых первое место по частоте занимает клапанный стеноз.
Нарушения гемодинамики зависят от степени стеноза, обусловливающего перегрузку правого желудочка. При незначительном стенозе жалобы отсутствуют, длительно сохраняется компенсация сердца, порок чаще выявляется в возрасте 10—15 лет.
При резком стенозе клинические симптомы порока появляются в раннем детском возрасте. Отмечаются жалобы на одышку, боли в области сердца, развивается относительная недостаточность трехстворчатого клапана с последующим возникновением симптомов правожелудочковой сердечной недостаточности. Границы сердца увеличены вправо. Во втором межреберье слева определяется систолическое дрожание, здесь же выслушивается систолический шум, интенсивность и продолжительность которого прямо пропорциональная степени стеноза; II тон над легочной артерией расщеплен, амплитуда его легочного компонента понижена.
На ФКГ шум имеет ромбовидную форму.
На ЭКГ отмечаются признаки гипертрофии правого желудочка, часто и правого предсердия.
Рентгенологически определяются увеличение правого желудочка, нередко и правого предсердия, постстенотическое расширение ствола легочной артерии. Легочный рисунок нормальный или обеднен.
Лечение. Хирургическое — вальвулотомия или баллонная вальвулопластика.
Тетрада Фалло
Сложный порок, состоит из 4 аномалий: стеноза выходного отдела правого желудочка, ДМЖП, декстропозиции аорты и гипертрофии правого желудочка. Нарушения гемодинамики определяются главным образом степенью стеноза легочной артерии и ДМЖП.
Выделяют следующие клинико-анатомические варианты порока: крайнюю, классическую и бледную формы.
У больных с крайней формой порока в связи с атрезией легочной артерии кровь в легкие поступает через ОАП или коллатерали.
При классической форме кровь из правого желудочка поступает в легочную артерию и аорту, при бледной форме происходит сброс крови слева направо, чем и объясняется отсутствие цианоза.
Клиническая картина порока в различные возрастные периоды имеет свои особенности. В первые месяцы жизни ребенка порок характеризуется бедностью симптоматики и дагностируется на основании грубого систолического шума в третьем-четвертом межреберье слева от грудины. С 4—6 мес начинается «критический период», который длится до 1,5—2 лет. У ребенка появляются одышка, цианоз, признаки отставания в физическом развитии, формируются симптомы «барабанных палочек» и «часовых стекол». В периферической крови увеличиваются уровень гемоглобина и число эритроцитов. При бледной форме тетрады Фалло одышка развивается позднее — к 5 годам.
Порок часто проявляется гипоксемическими (одышечно-цианотическими) приступами, которые возникают внезапно: ребенок становится беспокойным, резко усиливаются одышка и цианоз, аускультативно отмечают уменьшение интенсивности шума; возможна потеря сознания. В ряде случаев приступы сопровождаются развитием гемипареза. Приступы связаны со спазмом инфундибулярного отдела правого желудочка, они чаще отмечаются у больных с анемией и перенесших родовую травму. После 3—5 лет в большинстве случаев наступает улучшение состояния, что объясняется максимальной мобилизацией компенсаторных механизмов: развиваются полицитемия, коллатеральное кровообращение и др.
При тетраде Фалло в третьем-четвертом межреберье выслушивается грубый систолический шум, II тон над легочной артерией ослаблен. На ЭКГ регистрируются признаки гипертрофии правого желудочка.
На рентгенограммах сердце имеет форму деревянного башмачка, небольших размеров, сосудистый рисунок легких обеднен. При бледной форме порока размеры сердца обычно больше, увеличены за счет обоих желудочков, сосудистый рисунок легких усилен.
Наиболее тяжелым течением характеризуется крайняя форма тетрады Фалло, при которой резко выражен цианоз, дети значительно отстают в физическом развитии, систолический шум часто отсутствует.
Лечение. Хирургическое. Для профилактики приступов применяют анаприлин (обзидан) в дозе 1 мг/кг в сутки. С целью купирования одышечно-цианотических приступов парентерально вводят кордиамин, обзидан, переливают раствор глюкозы и эритроцитную массу (при анемии), при этом сердечные гликозиды противопоказаны.
Транспозиция магистральных сосудов полная
Встречается главным образом у детей раннего возраста (12—20 % от всех пороков). Дети умирают в возрасте до 1 года, в связи с чем у детей старшего возраста порок встречается значительно реже. При данном пороке аорта отходит от правого желудочка, а легочная артерия — от левого. Желудочки сердца и атриовентрикулярные клапаны сформированы правильно. Аорта чаще расположена впереди ствола легочной артерии. Неоксигенированная кровь из правого желудочка поступает в большой круг кровообращения, следствием чего является гипоксемия. В легкие из левого желудочка поступает кровь с повышенным содержанием кислорода.
Нарушения гемодинамики после рождения компенсируются сопутствующими пороками сердца: открытым овальным окном, ДМПП, ДМЖП и ОАП, без которых жизнь больных невозможна. Рано возникают симптомы ЗСН, которые быстро прогрессируют. Дети значительно отстают в физическом развитии, в относительно короткие сроки развиваются полицитемия, симптомы «барабанных палочек» и «часовых стекол». Границы сердца нормальные или слегка расширены, но в динамике они быстро увеличиваются. При аускультации шумы отсутствуют или выслушивается систолический шум, обусловленный ДМЖП или стенозом легочной артерии.
На ЭКГ чаще отмечаются признаки гипертрофии правого, реже обоих желудочков. На рентгенограммах форма сердца напоминает яйцо, лежащее на боку; вначале оно небольших размеров, затем развивается кардиомегалия. Легочный рисунок может быть нормальным, обедненным или усиленным.