Синдром Айкарди ( Aicardisyndrome )– это спорадическое заболевание, которое поражает преимущественно девочек и, предположительно, его причиной являются гетерозиготные мутации в Х-сцепленном гене.
Главные признаки заболевания включают триаду: инфантильные спазмы, агенезию мозолистого тела и характерные хориоретинальные вдавления. Дополнительные общие признаки включают: умеренную или глубокую умственную отсталость, гетеротопию серого вещества, аномалии извилин, дефекты позвонков и ребер.
На сегодняшний день нет описания характерных лицевых симптомов. Авторы исследовали 40 девочек с синдромом Айкарди и установили стойкие лицевые признаки, проявляющиеся у половины изученных пациентов, которые включали: выступающие резцы, вздернутый кончик носа, уменьшенный угол носовой перегородки и редкие, латерально расположенные (разбросанные) брови. Внешне видимая микрофтальмия наблюдалась у 10 из 40 пациентов (25 %). Различные кожные поражения (включающие невусы, кожные дивертикулы, гемангиомы, один гигантский меланоидный невус, и ( ранее удаленная ) ангиосаркома присутствовали у 8 из 40 пациентов (20%). Аномалии рук наблюдались у 3 из 40 (7,5 %) и включали компитодактилию, проксимальное расположение 1 пальца и гипоплазию 5-х пальцев рук.
Это исследование ясно наметило наличие характерных лицевых фенотипов синдрома Айкарди, описанных ранее отдельными исследователями. Авторы конкретизируют эти признаки: выступающая резцовая часть челюстных костей, уменьшенный угол носовой перегородки и сосудистые нарушения/опухоли, которые прибавляют модифицированные диагностические критерии в порядке улучшения возможности генетической диагностики синдрома Айкарди.
Синдром Айкарди впервые описан в середине 60-х годов, (Aicardietal., 1965) как генетическое неврологическое заболевание, поражающее преимущественно девочек (Hopkinsetal., 1979, Aicardi, 1999, VandenVeyver, 2002)
Исходное описание синдрома характеризовалось типичной триадой симптомов: агенезией мозолистого тела, типичными хориоретинальными углублениями (вдавлениями) и инфантильными спазмами ( Aicardietal.,1965,1969, Donnenfeldetal., 1989 ). Однако в большинстве установленных случаев становилось ясно, что и другие неврологические дефекты являются распространенными. На самом деле не все пораженные девочки имеют каждый признак из классической триады. Большая часть девочек с синдромом Айкарди имеют резкую задержку в интеллектуальном развитии, но в некоторых случаях имеется только средняя неспособность к обучению ( Meneezees et al.,1994 Yacoub et al.,2003 Chau et al., 2004, Matlary et al.,2004 ). Агенезия мозолистого тела не всегда присутствует, или она может быть частичной ( Donnenfeldetal.,1989, Aicardi,1999 ). Полимикрогирия или пахигирия, перивентрикулярная или внутрикорковая гетеротопия серого вещества, хориоидальные кисты и папилломы, вентрикуломегалия и внутримозговые кисты зачастую тоже присутствуют ( Aicardi,2005 ).
У большинства пораженных девочек после 3-х месячного возраста развивались инфантильные спазмы, и тяжелые эпилептические припадки сохранялись на протяжении всей жизни. На ЭЭГ демонстрировались асинхронные мультифокальные эпилептиформные очаги с вспышками супрессии и межполушарная диссоциация ( Fariello et al.,1977, Ohtsuki et al.,1981 ).
При синдроме Айкарди описано большое разнообразие дефектов развития глаз. Патогномоничные хориоретинальные углубления (вдавления) при синдроме Айкарди – белого, или желто-белого цвета, хорошо отграниченные, круглые депигментированные участки, расположенные на пигментном эпителии сетчатки, находящиеся под сосудистой оболочкой с различной плотностью депигментации на этих гранулах ( Donnenfeldetal .,1989, Carneyetal.,1993 ). Чувствительная сетчатка, лежащая над этими углублениями, обычно интактна, но может быть дезорганизована или полностью отсутствовать (DelPeroetal., 1986, Menezesetal.,1996). Другие описанные глазные аномалии включают микрофтальмию и колобомы, которые довольно типичны и вовлекают зрительный нерв, сосудистую оболочку и сетчатку, но радужка никогда не поражается.
Известны и другие врожденные дефекты при этом синдроме: полупозвонки и отсутствующие ребра, иногда приводящие к заметному сколиозу у трети пациентов ( Donnenfeldetal.,1989, Menezesetal.,1994).
Эти наблюдения большого процента нарушений, не входящих в классическую триаду, имеют значение для возможной корректировки критериев синдрома Айкарди (Aicardi 1999). Плагиоцефалия и лицевая асимметрия, порой с расщелиной верхней губы и неба (3%), тоже были описаны ( McPhersonandJones,1990 ), однако на сегодняшний день нет стойко идентифицированных лицевых фенотипов синдрома Айкарди.
В добавление показано, что могут увеличиваться случаи опухолей, более обычны папилломы сосудистого сплетения, но также описаны липомы, ангисаркомы, гепатобластомы, интерстициальный полипоз и эмбриональные кальциномы. ( Tanaka et al., 1985, Tagawa et al.,1989, Tsao et al.,1993,Trifiletti et al.,1995 ). Нет идентифицированного гена синдрома Айкарди, но серьезные изучения позволяют выдвигать гипотезу, что причиной синдрома Айкарди является мутация denovo на инактивированной Х-хромосоме ( VandenVeyver,2002 ). Почти все пораженные индивиды – девочки, и, предположительно, одна пара сестер ( Molinaetal 1989 ). Все описанные случаи спорадические. По крайней мере известно 6 пар близнецов, дискордантных по синдрому Айкарди, 5 из них – подтвержденные дизиготные, что исключает возможность пренатально-токсической этиологии и того, что это может быть дизрупцией ( TaggardandMenezes, 2000 ). Известно только три мальчика с подтвержденным диагнозом, имеющих кариотип 47, XXY ( Hopkinsetal., 1979, Aicardi,1999 ).
Поразительная вариабельность фенотипов при синдроме Айкарди может объясняться тем, что предполагаемый мутантный ген недостаточно инактивирует Х-хромосому (Wettke-SchaferandKantner,1983), однако некоторые публикации показали общую модель возможной случайной инактивации Х-хромосомы (Wieackeretal.,1985,Neidichetal.,1990,Hoagetal.,1997). Изучение установленной последовательности и делеции демонстрировало, что синдром Айкарди не является аллельным для синдрома Гольца ( Goltz syndrome ) или для микрофтальмии с линейными кожными дефектами ( Microphthalmia with Linear Skin Defects ) (VandenVeyveretal.,1998, VandenVeyver 2002). В настоящее время неизвестна локализация участка на Х-хромосоме, ответственного за проявление синдрома Айкарди.
Таблица № 1. Клинико-фенотипические признаки у 40 пациентов с синдромом Айкарди.
Пороки развития коры головного мозга ( чаще всего полимикрогирия )
Выступающие резцы со
вздернутым кончиком носа и редкими латеральными бровями
Хориоретинальные вдавления
Перивентрикулярная и субкортикальная гетеротопия
Пороки развития сосудов или злокачественные сосудистые опухоли
Инфантильные спазмы
Кисты вокруг III желудочка и/или сосудистого сплетения
Аномалии позвонков и ребер
Колобома диска зрительного нерва
Микрофтальмия, грубая полушарная асимметрия и ЭЭГ типа «расщепление мозга»
( “Split-brain” EEG )
Наличие двух классических симптомов и плюс два других признака достоверно свидетельствуют о синдроме Айкарди.
Авторы обследовали 40 индивидуумов с синдромом Айкарди и объективным образом определили узнаваемый лицевой профиль у большинства девочек с синдромом Айкарди. Лицевые дисморфии, описанные ранее в изолированных случаях, включали: «признаки дисморфии» (Meenezesetal., 1994): лицевую асимметрию (Aicardietal.,1969, Ohtsukietal.,1981), аномальные ушные раковины (Aicardietal.,1969, Ohtsukietal.,1981, Hamanoetal.,1991), уплощенную перегородку носа (Aicardietal.,1969, Ohtsukietal.,1981), гипертелоризм (WillisandRosman,1980), птоз, гипоплазию носа с развернутыми ноздрями, микрогнатию (Ohtsukietal.,1981). Об аномалиях верхних конечностей и стоп было также сообщено. Они включали: проксимально расположенные большие пальцы (Aicardietal.,1969, WillisandRosman,1980), асимметрию конечностей, единственную поперечную ладонную складку (WillisandRosman.,1980), неполное удвоение больших пальцев, синдактилию 2 и 3 пальцев стоп и гирсутизм (Ohtsukietal., 1981). Авторами впервые проведены систематизированные исследования лицевого фенотипа. Эти лицевые характеристики включали: выступающие резцы, развернутые ноздри с гипоплазией кончика носа и уменьшенным углом носовой перегородки, редкие латеральные брови. Авторы не подтвердили предыдущих сообщений о гипертелоризме и не наблюдали телекант. Дистанция между наружными углами глазной щели была меньше чем обычно, но с нормальной дистанцией между внутренними углами глаз, что вероятно походило на вторичную микрофтальмию, которая была у четверти наблюдаемых субъектов. Другие антропометрические замеры обнаруживают увеличенные ушные раковины и короткий фильтр, гипоплазию кистей со статистически значимым различием в размере по сравнению с общей популяцией.
Результатом хронической противосудорожной терапии может быть гиперплазия десен и огрубление черт лица. Однако авторы высказываю сомнение, что противосудорожное лечение ответственно за появление таких черт. Между тем отмечено, что резцы более резко начинают выступать с возрастом, хотя эти и другие лицевые черты видны уже и у очень маленьких девочек с синдромом Айкарди, а при независимом исследовании авторы собрали информацию об использовании широкого спектра действенных и новейших противосудорожных препаратов, таких как вигабартин, ламотриджин и топирамат, применение которых не могло быть ассоциировано с огрублением черт лица. Другие заболевания с хроническими и трудно поддающимися терапии судорогами, такие как синдром Ретта, синдром Ангельмана и некетотическая гиперглицинемия, не имеют сходных лицевых проявлений с синдромом Айкарди.
Авторы показали, что пальцевые, кожные и сосудистые аномалии встречались чаще, чем в общей популяции, но не подтвердили более высокую частоту незаращения губы и неба на предствленной серии больных (Umanskuetal.,1994). Наряду с тем, что аномалии кистей , такие как проксимально посаженные большие пальцы, удвоенные большие пальцы и синдактилия были установлены предварительно, частота их при синдроме Айкарди неизвестна.
Реберно-позвоночные аномалии встречаются у половины индивидуумов с синдромом Айкарди (Aicardietal.,1969, Hoytetal.,1978, Menezesetal.,1994).
Авторы указывают на повышенный процент сосудистых и пигментных кожных аномалий, что указывает на необходимость внимательного дерматологического наблюдения за сосудистыми и кожными поражениями у лиц с синдромом Айкарди. Меньшее число девочек, доказано имеющих синдром Айкарди, не имеют всех трех классических критериев синдрома. Это послужило основанием для модификации диагностических критериев которые были ранее предложены (Aicardi, 1999). В представленной авторами работе синдром Айкарди был чаще установлен офтальмологами и порой неврологами, т.е. клинические генетики не устанавливали первичный диагноз. Авторы надеются, что эти характеристики лицевых черт синдрома Айкарди будут совершенствовать диагностику синдрома Айкарди клиническими генетиками в особенности у индивидуумов, которые не имеют патогномоничных хориоретинальных вдавлений. Авторы рекомендуют использовать необходимые для оптимизации диагноза синдрома Айкарди диагностические черты в виде лицевых признаков: выступающих резцов со вздернутым кончиком носа и редкими латеральными бровями, равно как и сосудистые поражения ( табл.2).
Синдром Айкарди ( Aicardi syndrom) -это редкое генетическое заболевание, характеризующееся агенезией мозолистого тела, эпилептическими приступами по типу инфантильных спазмов с ранним дебютом, специфическими лакунарными изменениями на глазном дне, типичными изменениями на ЭЭГ (паттерн «расщепленного мозга»), задержкой психомоторного развития , а также лицевым дизморфизмом.
Мозолистое тело (большая спайка мозга)- представляет собой пласт нервных волокон, соединяющих кору двух полушарий большого мозга . Мозолистое тело формируется между 8-20 неделями гестации. После формирования мозолистое тело продолжает расти, увеличивается его длина и ширина. Перекрест волокон с проникновением из одного полушария в другое начинается на сроке 12 недель гестации.
Агенезия мозолистого тела относится к врожденным структурным нарушениям нейроонтогенеза. Она может быть изолированной, но чаще сочетается с другими врожденными аномалиями мозга - микрогирией, пахигирией, лиссэнцефалией, гидроцефалией. Клинические проявления агенезии мозолистого тела отличаются полиморфизмом: отмечается сочетание дизрафического статуса, умственной отсталости различной степени, эпилептических приступов, двигательных нарушений, и аномалии развития внутренних органов. Агенезия мозолистого тела может быть наследственно обусловлена и может являться результатом спонтанных мутации. Среди представленным в таблице вариантов агенезии мозолистого тела наиболее часто встречающимся и широко освещенным в литературе является синдром Айкарди.
Данные литературы о семейных случаях агенезии мозолистого тела (по I.Young)
В 1965 году французский невропатолог доктор Жан Айкарди описал 8 клинических случаев сочетания у детей инфантильных спазмов, агенезии мозолистого тела и пигментной дегенерации сетчатки. О таком клиническом случае уже сообщалось в 1949 году и еще тогда он был признан как случай заболевания, отличного от врожденной инфекции. Еще 7 пациентов было описано в 1969 году. И в 1972 году Dennis и Bower ввели термин - Синдром Айкарди. Дальнейшие исследования показали, что и другие аномалии развития вне классической триады симптомов, являются характерными для данного синдрома. Большинство детей имели задержку умственного развития различной степени, аномалии позвоночника и лицевой дизморфизм.
Важно сразу подчеркнуть, что синдром Айкарди не стоит путать с синдромом Aicardi-Goutieres ( или в литературе чаще также синдром Айкарди). Представляет собой прогрессирующую энцефалопатию с дебютом в раннем детском возрасте, сопровождающуюся кальцификацией базальных ганглиев, лейкодистрофией и хроническим лимфоцитозом СПЖ, с дебютом в раннем детском возрасте, аутосомно-рецессивным типом наследования. Описан в 1984 году. Клинические проявления включают микроцефалию, больные дети постепенно утрачивают приобретенные навыки, развивается раздражительность, наступает спастичность мышц, а так же возникают трудности при приеме пищи.
Распространенность.
Известно приблизительно о 500 случаях синдрома Айкарди во всем мире, особенно большое количество в Японии. Синдром встречается у детей с различной расовой принадлежностью. По недавно проведенным в Швеции исследованиям распространенность синдрома Айкарди составляет от 2 до 15 случаев на 100000 девочек.( В России аналогичные исследования к сожалению не проводились. ) Однако, учитывая фенотипическое разнообразие и диагностические трудности, многие случаи заболевания остаются недиагностированными. Это позволяет пересмотреть данные об истинной распространенности синдрома Айкарди в сторону увеличения, возможно, синдром Айкарди является более частой причиной задержки умственного развития и инфантильных спазмов у девочек, чем считается в настоящее время. На сегодняшний день считается, что заболеваемость синдромом Айкарди среди всех детей с инфантильными спазмами составляет всего около 2-4%.
Генетика.
Тип наследования синдрома Айкарди- доминантный сцепленный с Х-хромосомой, с предположительным локусом Хр22.3. Болеют только девочки. Трое мальчиков, которым был поставлен диагноз - синдром Айкарди имели генотип ХХY (47 хромосом)- синдром Кляйнфельтера [Hopkins и др.,1979, Aicardi 1999]. Для плода мужского пола с генотипом ХY синдром Айкарди является смертельным. За исключением одной пары сестер [ Molina, 1989] все описанные случаи являются спорадическими. Все случаи синдрома Айкарди, вероятно, связаны с новыми мутациями, так как ни одного случая передачи от матери к дочери гена, сцепленного с Х-хромосомой, отвечающего за развитие синдрома Айкарди, не зарегистрировано. Риск рождения в одной семье второго ребенка с синдромом Айкарди- менее 1%. Теоретически, риск передачи мутантного аллеля от женщины с синдромом Айкарди- 50%, однако плод мужского пола, носитель мутантного гена является нежизнеспособным. Таким образом, среди ожидаемого потомства 33%- здоровые девочки, 33%- здоровые мальчики и 33% - девочки с синдромом Айкарди.
Он и его семья пережили ужасный 1811-й голодный год в Мадриде, и эта жестокая проверка на выносливость стала для его жены Хосефы слишком тяжёлой. В июне 1812 года, она умерла, и 66-летний глухой старик остался один в своём доме.
Патогенез синдрома Айкарди в настоящее время неизвестен, связан с генетически детерминированными нарушениями этапа нейрональной миграции.
Патоморфология.
Патоморфологическое исследование головного мозга обычно демонстрирует множественные аномалии развития головного мозга, включающие полную или частичную агенезию мозолистого тела, гетеротопию коркового вещества мозга, аномалии строения извилин головного мозга, наиболее часто по типу микрогирии и гетеротопии, внутрижелудочковые кисты. При микроскопическом исследовании обычно обнаруживается нарушение клеточной архитектоники. При гистологическом исследовании сетчатки отмечается истончение всех слоев клеток, уменьшение числа и калибра сосудов, пигментная эктопия и гиперплазия пигментного эпителия.
Клиническая картина.
Дети, как правило, рождаются внешне здоровыми, с нормальным гестационным возрастом при рождении, без осложнений в пренатальном и интронатальном периоде, и развиваются по возрасту приблизительно до 2-5 (чаще 3) месяцев, когда чаще всего дебютируют инфантильные спазмы. Инфантильные спазмы - это тип эпилептических приступов, представляющих собой массивные миоклонические и(или) тонические, про- и(или) ретропульсивные, симметричные и(или) асимметричные, серийные и(или) изолированные спазмы аксиальной и конечностной мускулатуры. В 97% случаев при синдроме Айкарди наблюдаются флексорные инфантильные спазмы, которые могут быть атипичными, латерализованными. У 42% больных с синдромом Айкарди имеется сочетание инфантильных спазмов с другими видами эпилептических приступов, чаще с парциальными, реже с генерализованными тонико-клоническими. Итак, манифестными симптомами являются инфантильные спазмы либо реже парциальные приступы, дебют эпилептических приступов в 68% наблюдений приходится на первые три месяца жизни, в 23% случаев синдром дебютирует неонатальными судорогами в первый месяц жизни. Судорожные пароксизмы резистентны к проводимой противосудорожной терапии.
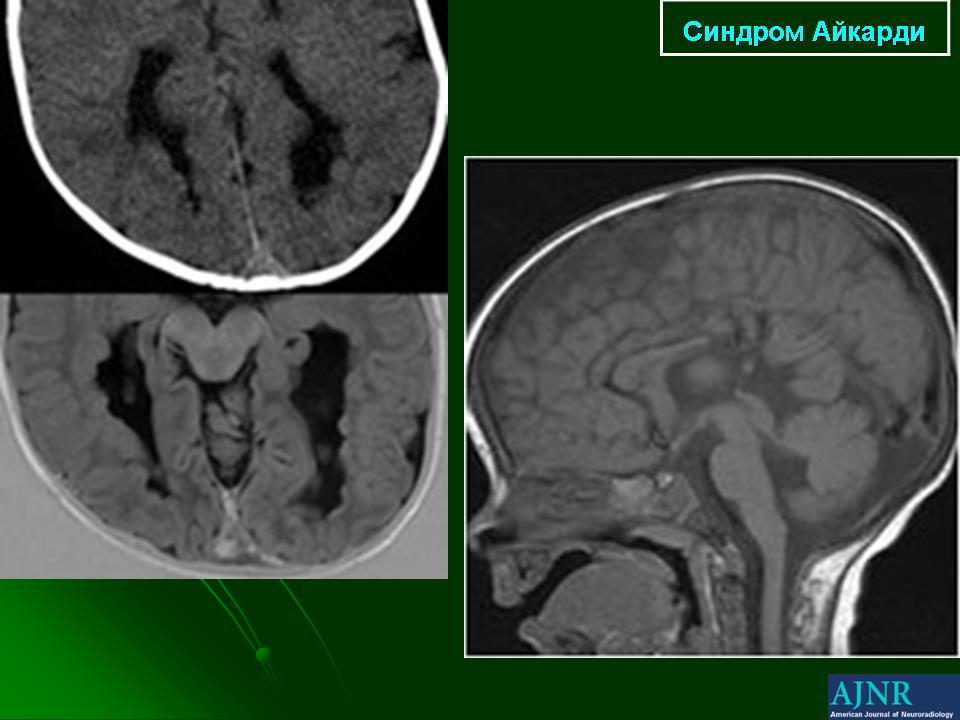
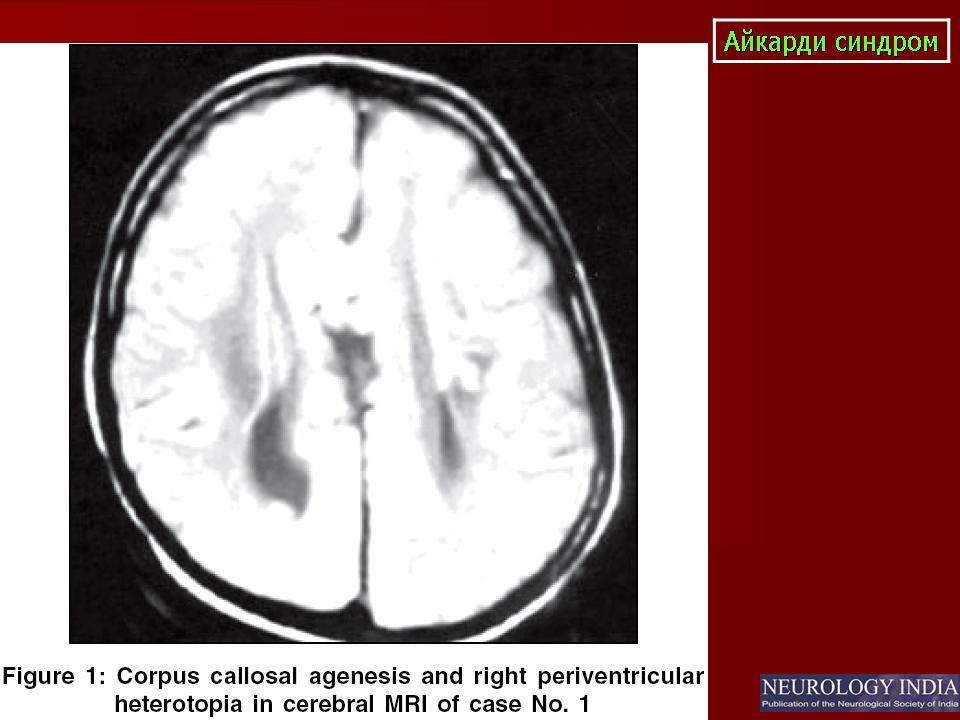
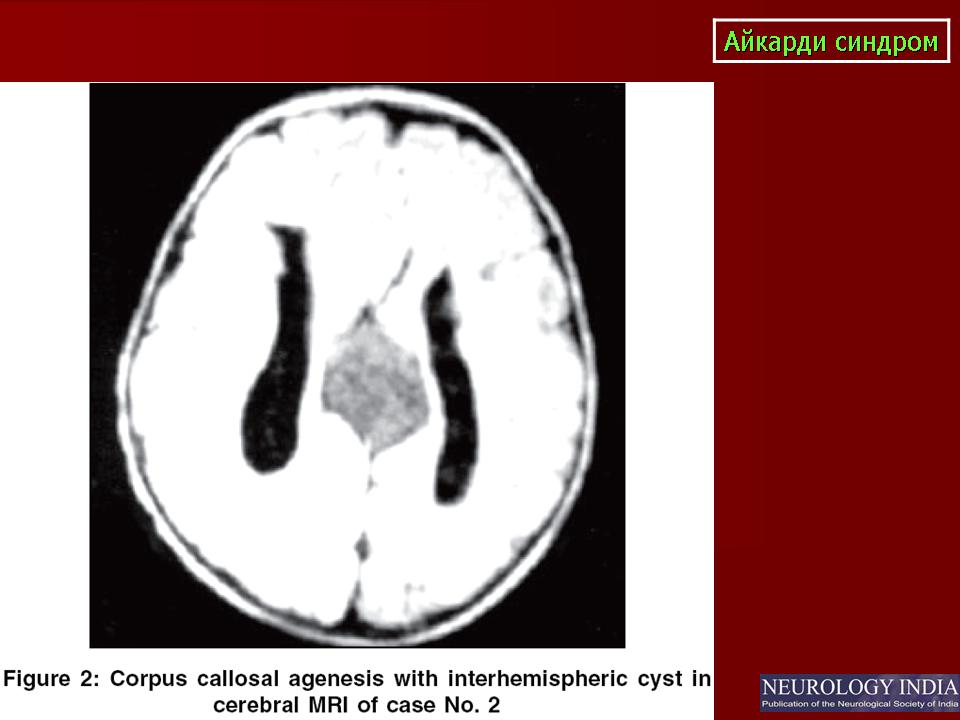
Большая часть девочек с синдромом Айкарди имеет резкую задержку психомоторного развития. Но описаны случаи незначительного снижения интеллекта и умеренной задержки развития [Chau et al 2004, Hatlary et al 2004, Prats Vinas etal 2005]. В неврологическом статусе часто отмечается -микроцефалия, мышечная гипотония, возможна односторонняя мышечная гипертония и спастичность, оживленные глубокие сухожильные рефлексы или геми- или тетрапарез [ Aicardi, 2005]. Агенезия мозолистого тела при синдроме Айкарди обычно тотальная, часто сочетается с гетеротопией коркового вещества мозга, атрофией коры, структурной асимметрией полушарий мозга, нормотензивной гидроцефалией, полимикрогирией или пахигирией, хориоидальными кистами и папилломами, вентрикуломегалией, внутримозговыми кистами, синдромом Денди-Уокера. Предполагается, что наличие комплекса аномалий нейрональной миграции даже более специфично для синдрома Айкарди, чем изолированная агенезия мозолистого тела [ Aicardi J., 1996].
Аномалии органа зрения: при синдроме Айкарди описано большое разнообразие аномалии развития глаз. Патогмоничным для данного синдрома является пигментный ретинит, проявляющийся различной степенью снижения остроты зрения (чаще довольно выраженной). Другие описанные аномалии глаз включают микрофтальмию, атрофию зрительного нерва, колобому, катаракту, которые могут быть одно- или двусторонними, или асимметричными.
Скелетные аномалии: отмечаются такие врожденные дефекты, как полупозвонки и отсутствующе ребра, иногда приводящие к выраженному сколиозу у трети пациентов [ Donnenfeld et al, 1989, Menezes et al, 1994].
Челюстно-лицевые аномалии: на сегодняшний день нет описания характерных лицевых симптомов при синдроме Айкарди, но наиболее часто встречающиеся, по данным авторов, наблюдавших 40 девочек с синдромом Айкарди, являются выступающие резцы, вздернутый кончик носа, уменьшенный угол носовой перегородки, редкие латерально расположенные брови ( признаки, проявившиеся у половины пациенток).Аномалии строения лица, описанные ранее в изолированных случаях, включают лицевую асимметрию [ Aicardi 1969], аномалии строения ушных раковин и уплощенную носовую перегородку[ Aicardi 1969, Ohtsuki et al, 1981], гипертелоризм [ Willis and Rosman, 1980], птоз, гипоплазию носа и микрогнатию [Ohtsuki et al, 1981], расщелину верхней губы и неба. Авторы также описали различные кожные поражения (включающее невусы, кожные дивертикулы, гемангиомы, ангиосаркомы) - у 20% пациенток. Пороки развития конечностей ( у 7,5%), включали каптодактилию, проксимальное расположение большого пальца, гипоплазию мизинцев, асимметрию конечностей, единственную поперечную ладонную складку, неполное удвоение большого пальца синдактилию 2 и 3 пальцев стоп.
Клинико-фенотипические признаки у 40 пациентов с синдромом Айкарди.
Желудочно-кишечный тракт: гастроезофагальный рефлюкс, запоры, диарея и трудности с кормлением.
У пациентов с синдромом Айкарди увеличена частота случаев опухолей. Чаще всего это папиллома сосудистых сплетений, так же описаны гемангиома, ангиосаркома, гепатобластома, полипоз кишечника, эмбриональные карциномы.
Рост. Темп роста замедляется в возрасте 7-10 лет и соответствует 5 центилю и ниже, увеличение веса также замедляется к этому возрасту до 25 центиля и ниже.
Эндокринная система. Возможно раннее наступление половой зрелости или задержка полового развития.
Критерии диагноза.
Классическое описание синдрома Айкарди включает триаду симптомов: агенезия мозолистого тела, инфантильные спазмы, пигментный ретинит. Однако, синдром Айкарди, как считается в настоящее время, является более сложным заболеванием, включающим дополнительные неврологические и экстраневральные симптомы. На этом основании были предложены модифицированные диагностические критерии синдрома Айкарди.
Для постановки диагноза синдром Айкарди необходимо присутствие всех трех классических симптомов или двух классических плюс, по крайней мере, два главных или дополнительных признака [Sutton et al 2005] .
Диагностика.
До настоящего времени не существует специального лабораторного диагностического теста или исследования, которое бы позволило поставить диагноз синдрома Айкарди. Для этого необходимо: неврологический осмотр, офтальмоскопия, ЭЭГ, МРТ с контрастом и/или без, рентгенограмма скелета.
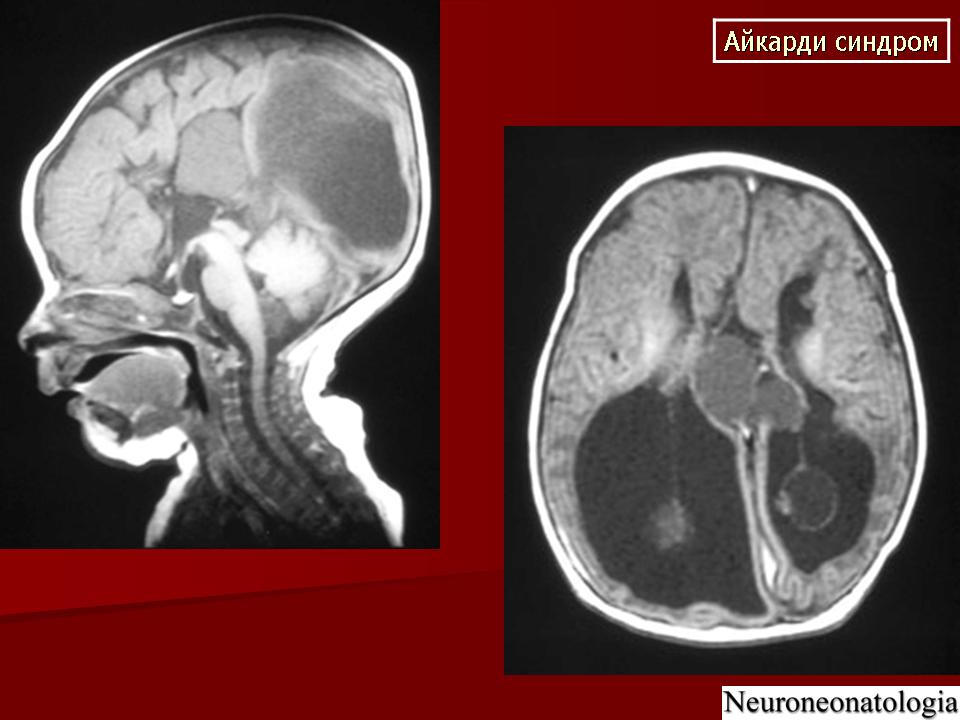
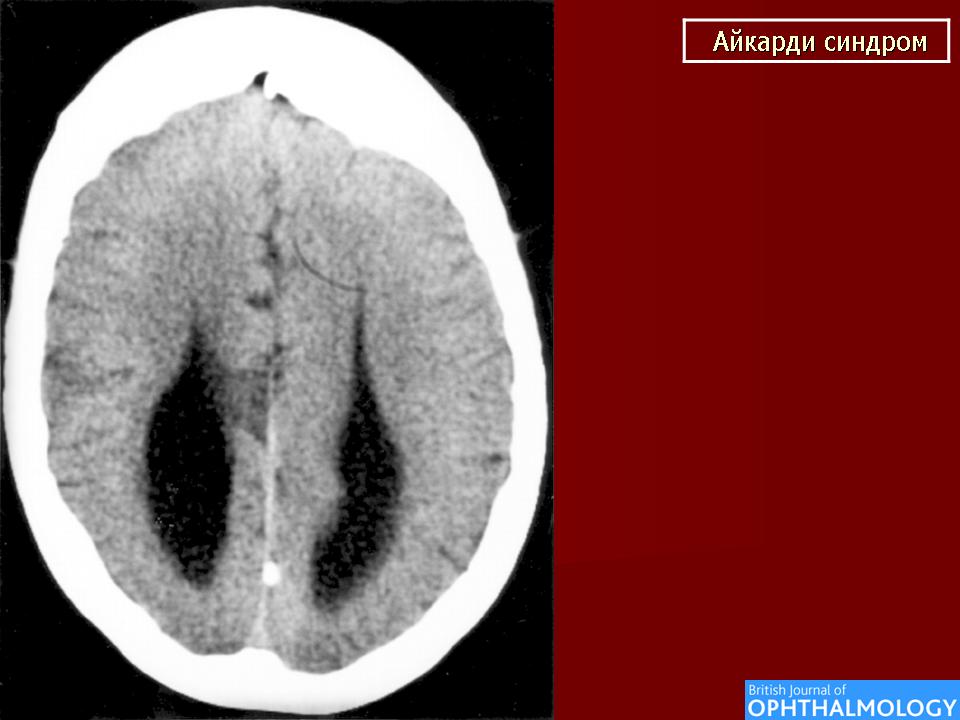
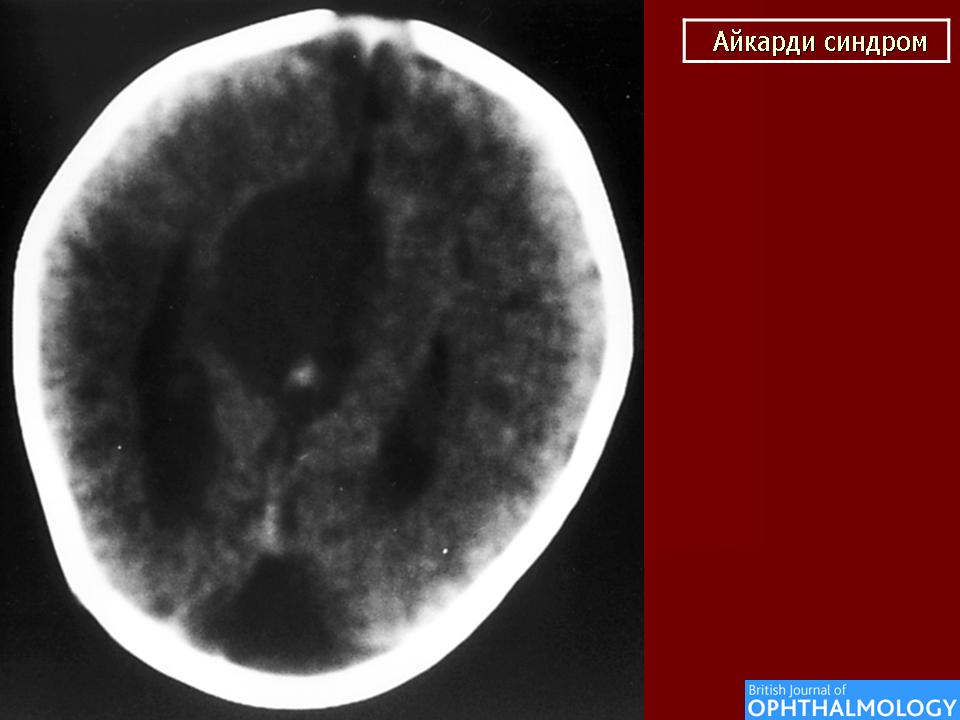
На МРТ можно обнаружить агенезию мозолистого тела, асимметрию полушарий коры, гетеротопию коркового вещества, внутримозговые кисты, папиллому сосудистых сплетений и тд.. Аксоны коры, которые в норме должны перекрещиваться, при его агенезии не формируются и соответственно не идентифицируются при нейровизуализации.
Агенезия мозолистого тела позволяет боковым желудочкам распространиться вверх, во фронтальное и париетальное белое вещество. Это состояние именуется верхней транслокацией боковых желудочков в лобно-теменные регионы мозга. Аналогичное смещение вверх претерпевает и III желудочек , что является одним из нейрорадиологических маркеров агенезии мозолистого тела. Увеличенный III желудочек, выдвигаясь вперед и вверх, раздвигает передние рога боковых желудочков, при сопутствующей гидроцефалии объем желудочков увеличивается, задние рога расширяются и изгибаются по направлению к средней линии (форма «ухвата»). Вероятно отсутствие поддерживающей функции мозолистого тела является основой для типичной черты агенезии мозолистого тела - расширения полушарий, III желудочка и Монроева отверстия.
Типичные изменения на ЭЭГ в виде гипсаритмии, характерные для инфантильных спазмов встречаются далеко не у всех больных. Наиболее характерные изменения приступной ЭЭГ заключаются во вспышках нерегулярных быстрых и медленных волн продолжительностью от 3 до 6 секунд, которые перемежаются некоторым уплощением основного ритма в течение 5-20 секунд, причем изменения не синхронизированы по полушариям. На ЭЭГ - феномен «расщепленного мозга» ("Split-brain"). Так как практически у половины (42%) пациентов инфантильные спазмы сочетаются с другими видами эпилептических приступов, то данные ЭЭГ могут быть противоречивы.
При офтальмоскопии обнаруживаются белого, или желто-белого цвета, хорошо отграниченные, круглые депигментированные участки.
Пренатальная диагностика.
Пренатальное молекулярно-генетическое исследование (ДНК диагностика) -пока что невозможно , так как ген точно не выявлен.Тем не менее, даже без ДНК-анализа при синдроме Айкарди возможна пренатальная диагностика с помощью ультразвукового исследования. Применяя методику ультразвукового сканирования плода во внутриутробном периоде на ранних сроках гестационного развития можно выявить агенезию мозолистого тела. Так же можно выявить некоторые другие аномалии, такие как внутримозговые кисты. Таким образом, ультразвуковой и , возможно, другие активно разрабатываемые в последние годы методы нейровизуализации (такие как МРТ плода) могут позволить заподозрить синдром Айкарди еще в пренатальном периоде.
Прогноз при синдроме Айкарди,как правило, серьезен в связи с выраженной умственной отсталостью и резистентным характером судорог. Часть детей (до 25%) погибает в первые годы жизни. Из выживших детей только 25% самостоятельно ходят и только 50% имеют навыки самообслуживания. Продолжительность жизни очень вариабельна, в зависимости от степени выраженности симптомов. Средняя продолжительность жизни по разным данным составляет от 8,3 до 18,5 лет. Но имеются сведения о женщине 32 лет с синдромом Айкарди, а так же 49 лет с умеренной формой синдрома.
Дифференциальный диагноз.
1)Агенезия мозолистого тела, изолированная или сочетанная с другими врожденными аномалиями.
2)Инфантильные спазмы (синдром Веста), которые встречаются у большинства девочек с синдромом Айкарди, но не являются специфичными для этого синдрома. Могут встречаться при других врожденных синдромах, нарушениях метаболизма и хромосомных болезнях.
3)Нарушения нейрональной миграции - полимикрогирия, гетеротопия, лиссэнцефалия, которые так же могут быть изолированно или как часть синдрома.

Синдром Айкарди ( Aicardi syndrome ) – это спорадическое заболевание, которое поражает преимущественно девочек и, предположительно, его причиной являются гетерозиготные мутации в Х-сцепленном гене.
Главные признаки заболевания включают триаду: инфантильные спазмы, агенезию мозолистого тела и характерные хориоретинальные вдавления. Дополнительные общие признаки включают: умеренную или глубокую умственную отсталость, гетеротопию серого вещества, аномалии извилин, дефекты позвонков и ребер.
На сегодняшний день нет описания характерных лицевых симптомов. Авторы исследовали 40 девочек с синдромом Айкарди и установили стойкие лицевые признаки, проявляющиеся у половины изученных пациентов, которые включали: выступающие резцы, вздернутый кончик носа, уменьшенный угол носовой перегородки и редкие, латерально расположенные (разбросанные) брови. Внешне видимая микрофтальмия наблюдалась у 10 из 40 пациентов (25 %). Различные кожные поражения (включающие невусы, кожные дивертикулы, гемангиомы, один гигантский меланоидный невус, и ( ранее удаленная ) ангиосаркома присутствовали у 8 из 40 пациентов (20%). Аномалии рук наблюдались у 3 из 40 (7,5 %) и включали компитодактилию, проксимальное расположение 1 пальца и гипоплазию 5-х пальцев рук.
Это исследование ясно наметило наличие характерных лицевых фенотипов синдрома Айкарди, описанных ранее отдельными исследователями. Авторы конкретизируют эти признаки: выступающая резцовая часть челюстных костей, уменьшенный угол носовой перегородки и сосудистые нарушения/опухоли, которые прибавляют модифицированные диагностические критерии в порядке улучшения возможности генетической диагностики синдрома Айкарди.
Синдром Айкарди впервые описан в середине 60-х годов, (Aicardi et al., 1965) как генетическое неврологическое заболевание, поражающее преимущественно девочек (Hopkins et al., 1979, Aicardi, 1999, Van den Veyver, 2002)
Исходное описание синдрома характеризовалось типичной триадой симптомов: агенезией мозолистого тела, типичными хориоретинальными углублениями (вдавлениями) и инфантильными спазмами ( Aicardi et al.,1965,1969, Donnenfeld et al., 1989 ). Однако в большинстве установленных случаев становилось ясно, что и другие неврологические дефекты являются распространенными. На самом деле не все пораженные девочки имеют каждый признак из классической триады. Большая часть девочек с синдромом Айкарди имеют резкую задержку в интеллектуальном развитии, но в некоторых случаях имеется только средняя неспособность к обучению ( Meneezees et al.,1994 Yacoub et al.,2003 Chau et al., 2004, Matlary et al.,2004 ). Агенезия мозолистого тела не всегда присутствует, или она может быть частичной ( Donnenfeld et al.,1989, Aicardi,1999 ). Полимикрогирия или пахигирия, перивентрикулярная или внутрикорковая гетеротопия серого вещества, хориоидальные кисты и папилломы, вентрикуломегалия и внутримозговые кисты зачастую тоже присутствуют ( Aicardi,2005 ).
У большинства пораженных девочек после 3-х месячного возраста развивались инфантильные спазмы, и тяжелые эпилептические припадки сохранялись на протяжении всей жизни. На ЭЭГ демонстрировались асинхронные мультифокальные эпилептиформные очаги с вспышками супрессии и межполушарная диссоциация ( Fariello et al.,1977, Ohtsuki et al.,1981 ).
При синдроме Айкарди описано большое разнообразие дефектов развития глаз. Патогномоничные хориоретинальные углубления (вдавления) при синдроме Айкарди – белого, или желто-белого цвета, хорошо отграниченные, круглые депигментированные участки, расположенные на пигментном эпителии сетчатки, находящиеся под сосудистой оболочкой с различной плотностью депигментации на этих гранулах ( Donnenfeld et al .,1989, Carney et al.,1993 ). Чувствительная сетчатка, лежащая над этими углублениями, обычно интактна, но может быть дезорганизована или полностью отсутствовать (Del Pero et al., 1986, Menezes et al.,1996). Другие описанные глазные аномалии включают микрофтальмию и колобомы, которые довольно типичны и вовлекают зрительный нерв, сосудистую оболочку и сетчатку, но радужка никогда не поражается.
Известны и другие врожденные дефекты при этом синдроме: полупозвонки и отсутствующие ребра, иногда приводящие к заметному сколиозу у трети пациентов ( Donnenfeld et al.,1989, Menezes et al.,1994).
Эти наблюдения большого процента нарушений, не входящих в классическую триаду, имеют значение для возможной корректировки критериев синдрома Айкарди (Aicardi 1999). Плагиоцефалия и лицевая асимметрия, порой с расщелиной верхней губы и неба (3%), тоже были описаны ( Mc Pherson and Jones,1990 ), однако на сегодняшний день нет стойко идентифицированных лицевых фенотипов синдрома Айкарди.
В добавление показано, что могут увеличиваться случаи опухолей, более обычны папилломы сосудистого сплетения, но также описаны липомы, ангисаркомы, гепатобластомы, интерстициальный полипоз и эмбриональные кальциномы. ( Tanaka et al., 1985, Tagawa et al.,1989, Tsao et al.,1993,Trifiletti et al.,1995 ). Нет идентифицированного гена синдрома Айкарди, но серьезные изучения позволяют выдвигать гипотезу, что причиной синдрома Айкарди является мутация de novo на инактивированной Х-хромосоме ( Van den Veyver,2002 ). Почти все пораженные индивиды – девочки, и, предположительно, одна пара сестер ( Molina et al 1989 ). Все описанные случаи спорадические. По крайней мере известно 6 пар близнецов, дискордантных по синдрому Айкарди, 5 из них – подтвержденные дизиготные, что исключает возможность пренатально-токсической этиологии и того, что это может быть дизрупцией ( Taggard and Menezes, 2000 ). Известно только три мальчика с подтвержденным диагнозом, имеющих кариотип 47, XXY ( Hopkins et al., 1979, Aicardi,1999 ).
Поразительная вариабельность фенотипов при синдроме Айкарди может объясняться тем, что предполагаемый мутантный ген недостаточно инактивирует Х-хромосому (Wettke-Schafer and Kantner,1983), однако некоторые публикации показали общую модель возможной случайной инактивации Х-хромосомы (Wieacker et al.,1985,Neidich et al.,1990,Hoag et al.,1997). Изучение установленной последовательности и делеции демонстрировало, что синдром Айкарди не является аллельным для синдрома Гольца ( Goltz syndrome ) или для микрофтальмии с линейными кожными дефектами ( Microphthalmia with Linear Skin Defects ) (Van den Veyver et al.,1998, Van den Veyver 2002). В настоящее время неизвестна локализация участка на Х-хромосоме, ответственного за проявление синдрома Айкарди.
Таблица № 1. Клинико-фенотипические признаки у 40 пациентов с синдромом Айкарди.
Признаки
Возраст 0 – 10 лет (n=31)
Возраст 10-16 лет (n=9)
Всего (n=40)
Выступающие резцы
20/31 (64,5%)
6/9 (66,7%)
26/40 (65%)
Редкие латеральные брови
12/31 (38,7%)
5/9 (55,6%)
17/40 (42,5%)
Кожные поражения
7/31 (22,6%)
2/9 (22,2%)
9/40 (22,5%)
Порок развития верхних конечностей
3/31 (9,7%)
0/9 (0%)
3/40 (7.5%)
Таблица № 2. Модифицированные диагностические критерии синдрома Айкарди.
Классическая триада
Главные признаки
Вспомогательные признаки
Агенезия мозолистого тела
Пороки развития коры головного мозга ( чаще всего полимикрогирия )
Выступающие резцы со
вздернутым кончиком носа и редкими латеральными бровями
Хориоретинальные вдавления
Перивентрикулярная и субкортикальная гетеротопия
Пороки развития сосудов или злокачественные сосудистые опухоли
Инфантильные спазмы
Кисты вокруг III желудочка и/или сосудистого сплетения
Аномалии позвонков и ребер
Колобома диска зрительного нерва
Микрофтальмия, грубая полушарная асимметрия и ЭЭГ типа «расщепление мозга»
( “Split-brain” EEG )
Наличие двух классических симптомов и плюс два других признака достоверно свидетельствуют о синдроме Айкарди.
Авторы обследовали 40 индивидуумов с синдромом Айкарди и объективным образом определили узнаваемый лицевой профиль у большинства девочек с синдромом Айкарди. Лицевые дисморфии, описанные ранее в изолированных случаях, включали: «признаки дисморфии» (Meenezes et al., 1994): лицевую асимметрию (Aicardi et al.,1969, Ohtsuki et al.,1981), аномальные ушные раковины (Aicardi et al.,1969, Ohtsuki et al.,1981, Hamano et al.,1991), уплощенную перегородку носа (Aicardi et al.,1969, Ohtsuki et al.,1981), гипертелоризм (Willis and Rosman,1980), птоз, гипоплазию носа с развернутыми ноздрями, микрогнатию (Ohtsuki et al.,1981). Об аномалиях верхних конечностей и стоп было также сообщено. Они включали: проксимально расположенные большие пальцы (Aicardi et al.,1969, Willis and Rosman,1980), асимметрию конечностей, единственную поперечную ладонную складку (Willis and Rosman.,1980), неполное удвоение больших пальцев, синдактилию 2 и 3 пальцев стоп и гирсутизм (Ohtsuki et al., 1981). Авторами впервые проведены систематизированные исследования лицевого фенотипа. Эти лицевые характеристики включали: выступающие резцы, развернутые ноздри с гипоплазией кончика носа и уменьшенным углом носовой перегородки, редкие латеральные брови. Авторы не подтвердили предыдущих сообщений о гипертелоризме и не наблюдали телекант. Дистанция между наружными углами глазной щели была меньше чем обычно, но с нормальной дистанцией между внутренними углами глаз, что вероятно походило на вторичную микрофтальмию, которая была у четверти наблюдаемых субъектов. Другие антропометрические замеры обнаруживают увеличенные ушные раковины и короткий фильтр, гипоплазию кистей со статистически значимым различием в размере по сравнению с общей популяцией.
Результатом хронической противосудорожной терапии может быть гиперплазия десен и огрубление черт лица. Однако авторы высказываю сомнение, что противосудорожное лечение ответственно за появление таких черт. Между тем отмечено, что резцы более резко начинают выступать с возрастом, хотя эти и другие лицевые черты видны уже и у очень маленьких девочек с синдромом Айкарди, а при независимом исследовании авторы собрали информацию об использовании широкого спектра действенных и новейших противосудорожных препаратов, таких как вигабартин, ламотриджин и топирамат, применение которых не могло быть ассоциировано с огрублением черт лица. Другие заболевания с хроническими и трудно поддающимися терапии судорогами, такие как синдром Ретта, синдром Ангельмана и некетотическая гиперглицинемия, не имеют сходных лицевых проявлений с синдромом Айкарди.
Авторы показали, что пальцевые, кожные и сосудистые аномалии встречались чаще, чем в общей популяции, но не подтвердили более высокую частоту незаращения губы и неба на предствленной серии больных (Umansku et al.,1994). Наряду с тем, что аномалии кистей , такие как проксимально посаженные большие пальцы, удвоенные большие пальцы и синдактилия были установлены предварительно, частота их при синдроме Айкарди неизвестна.
Реберно-позвоночные аномалии встречаются у половины индивидуумов с синдромом Айкарди (Aicardi et al.,1969, Hoyt et al.,1978, Menezes et al.,1994).
Авторы указывают на повышенный процент сосудистых и пигментных кожных аномалий, что указывает на необходимость внимательного дерматологического наблюдения за сосудистыми и кожными поражениями у лиц с синдромом Айкарди. Меньшее число девочек, доказано имеющих синдром Айкарди, не имеют всех трех классических критериев синдрома. Это послужило основанием для модификации диагностических критериев которые были ранее предложены (Aicardi, 1999). В представленной авторами работе синдром Айкарди был чаще установлен офтальмологами и порой неврологами, т.е. клинические генетики не устанавливали первичный диагноз. Авторы надеются, что эти характеристики лицевых черт синдрома Айкарди будут совершенствовать диагностику синдрома Айкарди клиническими генетиками в особенности у индивидуумов, которые не имеют патогномоничных хориоретинальных вдавлений. Авторы рекомендуют использовать необходимые для оптимизации диагноза синдрома Айкарди диагностические черты в виде лицевых признаков: выступающих резцов со вздернутым кончиком носа и редкими латеральными бровями, равно как и сосудистые поражения ( табл.2).
Девочка страдает синдромом Айкарди
Синдром Айкарди ( Aicardi syndrom) -это редкое генетическое заболевание, характеризующееся агенезией мозолистого тела, эпилептическими приступами по типу инфантильных спазмов с ранним дебютом, специфическими лакунарными изменениями на глазном дне, типичными изменениями на ЭЭГ (паттерн «расщепленного мозга»), задержкой психомоторного развития , а также лицевым дизморфизмом.
Мозолистое тело (большая спайка мозга)- представляет собой пласт нервных волокон, соединяющих кору двух полушарий большого мозга . Мозолистое тело формируется между 8-20 неделями гестации. После формирования мозолистое тело продолжает расти, увеличивается его длина и ширина. Перекрест волокон с проникновением из одного полушария в другое начинается на сроке 12 недель гестации.
Агенезия мозолистого тела относится к врожденным структурным нарушениям нейроонтогенеза. Она может быть изолированной, но чаще сочетается с другими врожденными аномалиями мозга - микрогирией, пахигирией, лиссэнцефалией, гидроцефалией. Клинические проявления агенезии мозолистого тела отличаются полиморфизмом: отмечается сочетание дизрафического статуса, умственной отсталости различной степени, эпилептических приступов, двигательных нарушений, и аномалии развития внутренних органов. Агенезия мозолистого тела может быть наследственно обусловлена и может являться результатом спонтанных мутации. Среди представленным в таблице вариантов агенезии мозолистого тела наиболее часто встречающимся и широко освещенным в литературе является синдром Айкарди.
Данные литературы о семейных случаях агенезии мозолистого тела (по I.Young)
Историческая справка.
В 1965 году французский невропатолог доктор Жан Айкарди описал 8 клинических случаев сочетания у детей инфантильных спазмов, агенезии мозолистого тела и пигментной дегенерации сетчатки. О таком клиническом случае уже сообщалось в 1949 году и еще тогда он был признан как случай заболевания, отличного от врожденной инфекции. Еще 7 пациентов было описано в 1969 году. И в 1972 году Dennis и Bower ввели термин - Синдром Айкарди. Дальнейшие исследования показали, что и другие аномалии развития вне классической триады симптомов, являются характерными для данного синдрома. Большинство детей имели задержку умственного развития различной степени, аномалии позвоночника и лицевой дизморфизм.
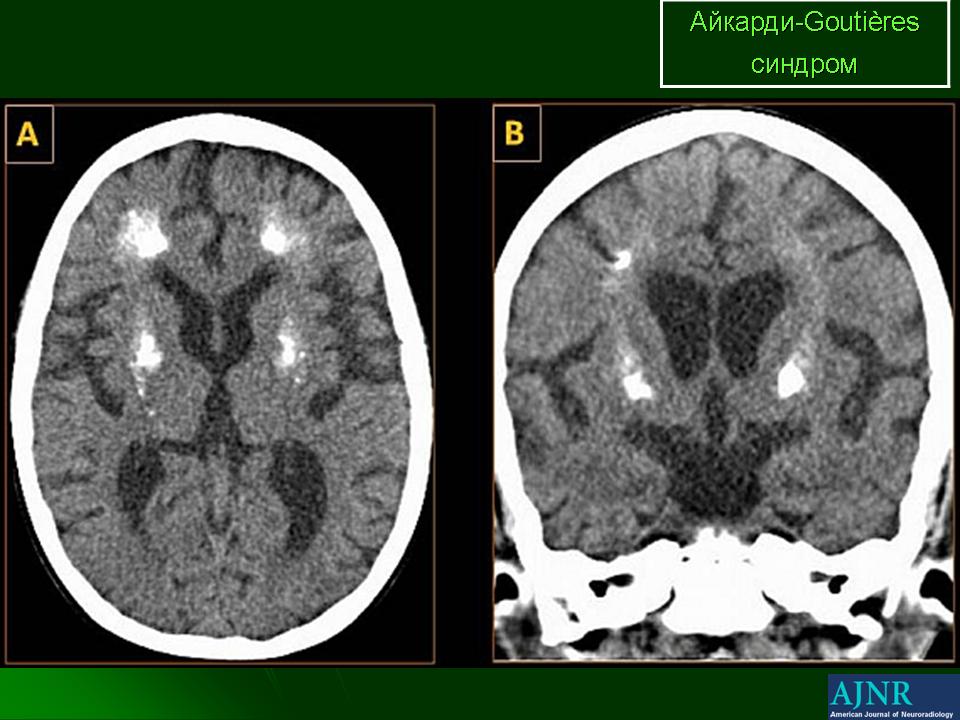
Важно сразу подчеркнуть, что синдром Айкарди не стоит путать с синдромом Aicardi-Goutieres ( или в литературе чаще также синдром Айкарди). Представляет собой прогрессирующую энцефалопатию с дебютом в раннем детском возрасте, сопровождающуюся кальцификацией базальных ганглиев, лейкодистрофией и хроническим лимфоцитозом СПЖ, с дебютом в раннем детском возрасте, аутосомно-рецессивным типом наследования. Описан в 1984 году. Клинические проявления включают микроцефалию, больные дети постепенно утрачивают приобретенные навыки, развивается раздражительность, наступает спастичность мышц, а так же возникают трудности при приеме пищи.
Распространенность.
Известно приблизительно о 500 случаях синдрома Айкарди во всем мире, особенно большое количество в Японии. Синдром встречается у детей с различной расовой принадлежностью. По недавно проведенным в Швеции исследованиям распространенность синдрома Айкарди составляет от 2 до 15 случаев на 100000 девочек.( В России аналогичные исследования к сожалению не проводились. ) Однако, учитывая фенотипическое разнообразие и диагностические трудности, многие случаи заболевания остаются недиагностированными. Это позволяет пересмотреть данные об истинной распространенности синдрома Айкарди в сторону увеличения, возможно, синдром Айкарди является более частой причиной задержки умственного развития и инфантильных спазмов у девочек, чем считается в настоящее время. На сегодняшний день считается, что заболеваемость синдромом Айкарди среди всех детей с инфантильными спазмами составляет всего около 2-4%.
Генетика.
Тип наследования синдрома Айкарди- доминантный сцепленный с Х-хромосомой, с предположительным локусом Хр22.3. Болеют только девочки. Трое мальчиков, которым был поставлен диагноз - синдром Айкарди имели генотип ХХY (47 хромосом)- синдром Кляйнфельтера [Hopkins и др.,1979, Aicardi 1999]. Для плода мужского пола с генотипом ХY синдром Айкарди является смертельным. За исключением одной пары сестер [ Molina, 1989] все описанные случаи являются спорадическими. Все случаи синдрома Айкарди, вероятно, связаны с новыми мутациями, так как ни одного случая передачи от матери к дочери гена, сцепленного с Х-хромосомой, отвечающего за развитие синдрома Айкарди, не зарегистрировано. Риск рождения в одной семье второго ребенка с синдромом Айкарди- менее 1%. Теоретически, риск передачи мутантного аллеля от женщины с синдромом Айкарди- 50%, однако плод мужского пола, носитель мутантного гена является нежизнеспособным. Таким образом, среди ожидаемого потомства 33%- здоровые девочки, 33%- здоровые мальчики и 33% - девочки с синдромом Айкарди.
Он и его семья пережили ужасный 1811-й голодный год в Мадриде, и эта жестокая проверка на выносливость стала для его жены Хосефы слишком тяжёлой. В июне 1812 года, она умерла, и 66-летний глухой старик остался один в своём доме.
Патогенез синдрома Айкарди в настоящее время неизвестен, связан с генетически детерминированными нарушениями этапа нейрональной миграции.
Патоморфология.
Патоморфологическое исследование головного мозга обычно демонстрирует множественные аномалии развития головного мозга, включающие полную или частичную агенезию мозолистого тела, гетеротопию коркового вещества мозга, аномалии строения извилин головного мозга, наиболее часто по типу микрогирии и гетеротопии, внутрижелудочковые кисты. При микроскопическом исследовании обычно обнаруживается нарушение клеточной архитектоники. При гистологическом исследовании сетчатки отмечается истончение всех слоев клеток, уменьшение числа и калибра сосудов, пигментная эктопия и гиперплазия пигментного эпителия.
Клиническая картина.
Дети, как правило, рождаются внешне здоровыми, с нормальным гестационным возрастом при рождении, без осложнений в пренатальном и интронатальном периоде, и развиваются по возрасту приблизительно до 2-5 (чаще 3) месяцев, когда чаще всего дебютируют инфантильные спазмы. Инфантильные спазмы - это тип эпилептических приступов, представляющих собой массивные миоклонические и(или) тонические, про- и(или) ретропульсивные, симметричные и(или) асимметричные, серийные и(или) изолированные спазмы аксиальной и конечностной мускулатуры. В 97% случаев при синдроме Айкарди наблюдаются флексорные инфантильные спазмы, которые могут быть атипичными, латерализованными. У 42% больных с синдромом Айкарди имеется сочетание инфантильных спазмов с другими видами эпилептических приступов, чаще с парциальными, реже с генерализованными тонико-клоническими. Итак, манифестными симптомами являются инфантильные спазмы либо реже парциальные приступы, дебют эпилептических приступов в 68% наблюдений приходится на первые три месяца жизни, в 23% случаев синдром дебютирует неонатальными судорогами в первый месяц жизни. Судорожные пароксизмы резистентны к проводимой противосудорожной терапии.
Большая часть девочек с синдромом Айкарди имеет резкую задержку психомоторного развития. Но описаны случаи незначительного снижения интеллекта и умеренной задержки развития [Chau et al 2004, Hatlary et al 2004, Prats Vinas etal 2005]. В неврологическом статусе часто отмечается -микроцефалия, мышечная гипотония, возможна односторонняя мышечная гипертония и спастичность, оживленные глубокие сухожильные рефлексы или геми- или тетрапарез [ Aicardi, 2005]. Агенезия мозолистого тела при синдроме Айкарди обычно тотальная, часто сочетается с гетеротопией коркового вещества мозга, атрофией коры, структурной асимметрией полушарий мозга, нормотензивной гидроцефалией, полимикрогирией или пахигирией, хориоидальными кистами и папилломами, вентрикуломегалией, внутримозговыми кистами, синдромом Денди-Уокера. Предполагается, что наличие комплекса аномалий нейрональной миграции даже более специфично для синдрома Айкарди, чем изолированная агенезия мозолистого тела [ Aicardi J., 1996].
Аномалии органа зрения: при синдроме Айкарди описано большое разнообразие аномалии развития глаз. Патогмоничным для данного синдрома является пигментный ретинит, проявляющийся различной степенью снижения остроты зрения (чаще довольно выраженной). Другие описанные аномалии глаз включают микрофтальмию, атрофию зрительного нерва, колобому, катаракту, которые могут быть одно- или двусторонними, или асимметричными.
Скелетные аномалии: отмечаются такие врожденные дефекты, как полупозвонки и отсутствующе ребра, иногда приводящие к выраженному сколиозу у трети пациентов [ Donnenfeld et al, 1989, Menezes et al, 1994].
Челюстно-лицевые аномалии: на сегодняшний день нет описания характерных лицевых симптомов при синдроме Айкарди, но наиболее часто встречающиеся, по данным авторов, наблюдавших 40 девочек с синдромом Айкарди, являются выступающие резцы, вздернутый кончик носа, уменьшенный угол носовой перегородки, редкие латерально расположенные брови ( признаки, проявившиеся у половины пациенток).Аномалии строения лица, описанные ранее в изолированных случаях, включают лицевую асимметрию [ Aicardi 1969], аномалии строения ушных раковин и уплощенную носовую перегородку[ Aicardi 1969, Ohtsuki et al, 1981], гипертелоризм [ Willis and Rosman, 1980], птоз, гипоплазию носа и микрогнатию [Ohtsuki et al, 1981], расщелину верхней губы и неба. Авторы также описали различные кожные поражения (включающее невусы, кожные дивертикулы, гемангиомы, ангиосаркомы) - у 20% пациенток. Пороки развития конечностей ( у 7,5%), включали каптодактилию, проксимальное расположение большого пальца, гипоплазию мизинцев, асимметрию конечностей, единственную поперечную ладонную складку, неполное удвоение большого пальца синдактилию 2 и 3 пальцев стоп.
Клинико-фенотипические признаки у 40 пациентов с синдромом Айкарди.
Желудочно-кишечный тракт: гастроезофагальный рефлюкс, запоры, диарея и трудности с кормлением.
У пациентов с синдромом Айкарди увеличена частота случаев опухолей. Чаще всего это папиллома сосудистых сплетений, так же описаны гемангиома, ангиосаркома, гепатобластома, полипоз кишечника, эмбриональные карциномы.
Рост. Темп роста замедляется в возрасте 7-10 лет и соответствует 5 центилю и ниже, увеличение веса также замедляется к этому возрасту до 25 центиля и ниже.
Эндокринная система. Возможно раннее наступление половой зрелости или задержка полового развития.
Критерии диагноза.
Классическое описание синдрома Айкарди включает триаду симптомов: агенезия мозолистого тела, инфантильные спазмы, пигментный ретинит. Однако, синдром Айкарди, как считается в настоящее время, является более сложным заболеванием, включающим дополнительные неврологические и экстраневральные симптомы. На этом основании были предложены модифицированные диагностические критерии синдрома Айкарди.
Модифицированные диагностические критерии синдрома Айкарди.
Для постановки диагноза синдром Айкарди необходимо присутствие всех трех классических симптомов или двух классических плюс, по крайней мере, два главных или дополнительных признака [Sutton et al 2005] .
Диагностика.
До настоящего времени не существует специального лабораторного диагностического теста или исследования, которое бы позволило поставить диагноз синдрома Айкарди. Для этого необходимо: неврологический осмотр, офтальмоскопия, ЭЭГ, МРТ с контрастом и/или без, рентгенограмма скелета.
На МРТ можно обнаружить агенезию мозолистого тела, асимметрию полушарий коры, гетеротопию коркового вещества, внутримозговые кисты, папиллому сосудистых сплетений и тд.. Аксоны коры, которые в норме должны перекрещиваться, при его агенезии не формируются и соответственно не идентифицируются при нейровизуализации.
Агенезия мозолистого тела позволяет боковым желудочкам распространиться вверх, во фронтальное и париетальное белое вещество. Это состояние именуется верхней транслокацией боковых желудочков в лобно-теменные регионы мозга. Аналогичное смещение вверх претерпевает и III желудочек , что является одним из нейрорадиологических маркеров агенезии мозолистого тела. Увеличенный III желудочек, выдвигаясь вперед и вверх, раздвигает передние рога боковых желудочков, при сопутствующей гидроцефалии объем желудочков увеличивается, задние рога расширяются и изгибаются по направлению к средней линии (форма «ухвата»). Вероятно отсутствие поддерживающей функции мозолистого тела является основой для типичной черты агенезии мозолистого тела - расширения полушарий, III желудочка и Монроева отверстия.
Типичные изменения на ЭЭГ в виде гипсаритмии, характерные для инфантильных спазмов встречаются далеко не у всех больных. Наиболее характерные изменения приступной ЭЭГ заключаются во вспышках нерегулярных быстрых и медленных волн продолжительностью от 3 до 6 секунд, которые перемежаются некоторым уплощением основного ритма в течение 5-20 секунд, причем изменения не синхронизированы по полушариям. На ЭЭГ - феномен «расщепленного мозга» ("Split-brain"). Так как практически у половины (42%) пациентов инфантильные спазмы сочетаются с другими видами эпилептических приступов, то данные ЭЭГ могут быть противоречивы.
При офтальмоскопии обнаруживаются белого, или желто-белого цвета, хорошо отграниченные, круглые депигментированные участки.
Пренатальная диагностика.
Пренатальное молекулярно-генетическое исследование (ДНК диагностика) -пока что невозможно , так как ген точно не выявлен.Тем не менее, даже без ДНК-анализа при синдроме Айкарди возможна пренатальная диагностика с помощью ультразвукового исследования. Применяя методику ультразвукового сканирования плода во внутриутробном периоде на ранних сроках гестационного развития можно выявить агенезию мозолистого тела. Так же можно выявить некоторые другие аномалии, такие как внутримозговые кисты. Таким образом, ультразвуковой и , возможно, другие активно разрабатываемые в последние годы методы нейровизуализации (такие как МРТ плода) могут позволить заподозрить синдром Айкарди еще в пренатальном периоде.
Прогноз при синдроме Айкарди,как правило, серьезен в связи с выраженной умственной отсталостью и резистентным характером судорог. Часть детей (до 25%) погибает в первые годы жизни. Из выживших детей только 25% самостоятельно ходят и только 50% имеют навыки самообслуживания. Продолжительность жизни очень вариабельна, в зависимости от степени выраженности симптомов. Средняя продолжительность жизни по разным данным составляет от 8,3 до 18,5 лет. Но имеются сведения о женщине 32 лет с синдромом Айкарди, а так же 49 лет с умеренной формой синдрома.
Дифференциальный диагноз.
1)Агенезия мозолистого тела, изолированная или сочетанная с другими врожденными аномалиями.
2)Инфантильные спазмы (синдром Веста), которые встречаются у большинства девочек с синдромом Айкарди, но не являются специфичными для этого синдрома. Могут встречаться при других врожденных синдромах, нарушениях метаболизма и хромосомных болезнях.
3)Нарушения нейрональной миграции - полимикрогирия, гетеротопия, лиссэнцефалия, которые так же могут быть изолированно или как часть синдрома.
https://radiopaedia.org/articles/aicardi-syndrome