Мукополисахаридоз I типа (синдром Гурлер) описан G. Hurler в 1919 г. Наследуется аутосомно-рецессивно. В основе патогенеза лежит дефицит фермента а — L-идуронидазы, который приводит к нарушению катаболизма дерматансульфата и гепарансульфата, накапливающихся в клетках и экскретируемых с мочой.
При патоморфологическом исследовании выявляется отложение мукополисахаридов в виде зернистой массы в паренхиме печени, ретикулярных клетках селезенки, эпителии почечных канальцев; сетчатке, склере, роговице глаз; нервных клетках, периферических ганглиях, в интиме коронарных артерий, бронхиальных и других хрящах. Отмечается внутренняя гидроцефалия, обусловленная, по-видимому, отложением мукополисахаридов в мозговых оболочках и нарушением их проницаемости. Определяются очаги демиелинизации. Реактивный фиброз эндокарда связан с отложением мукополисахаридов в клапанах.
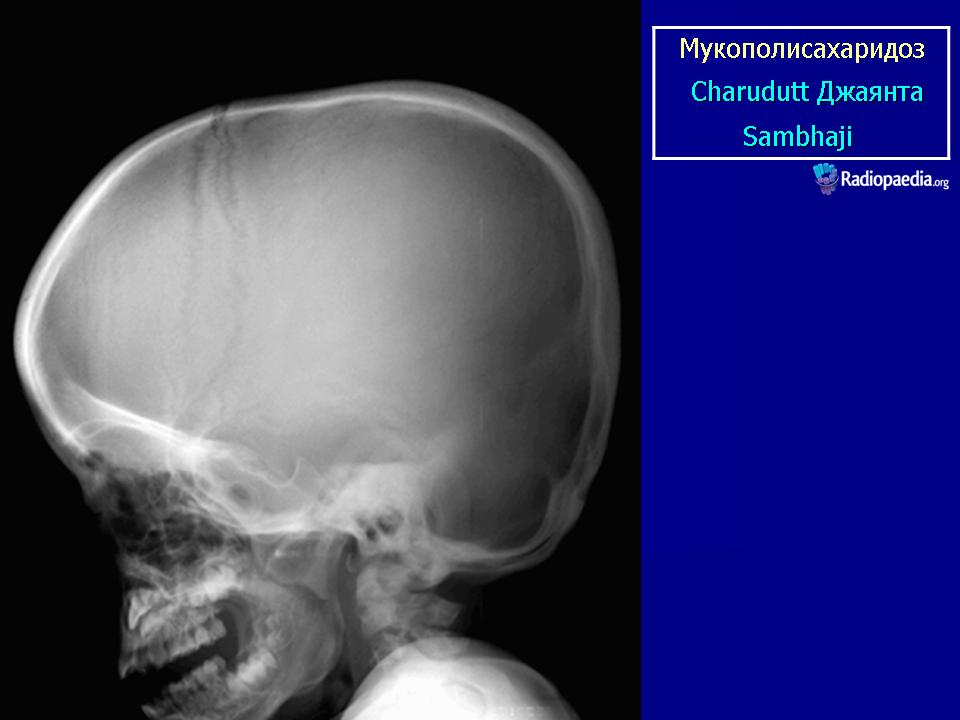
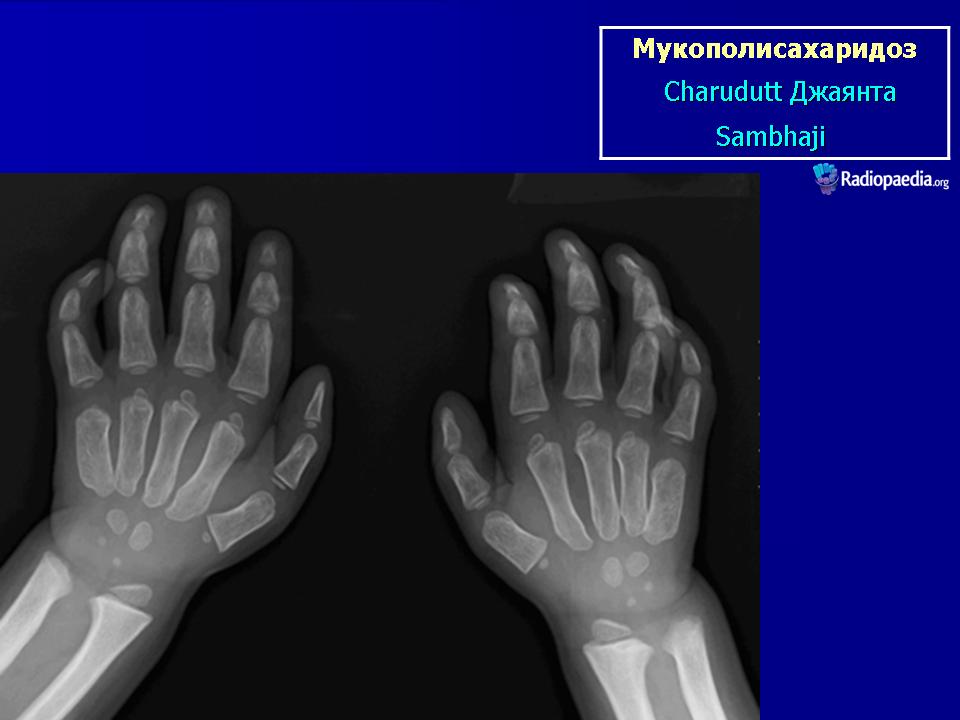
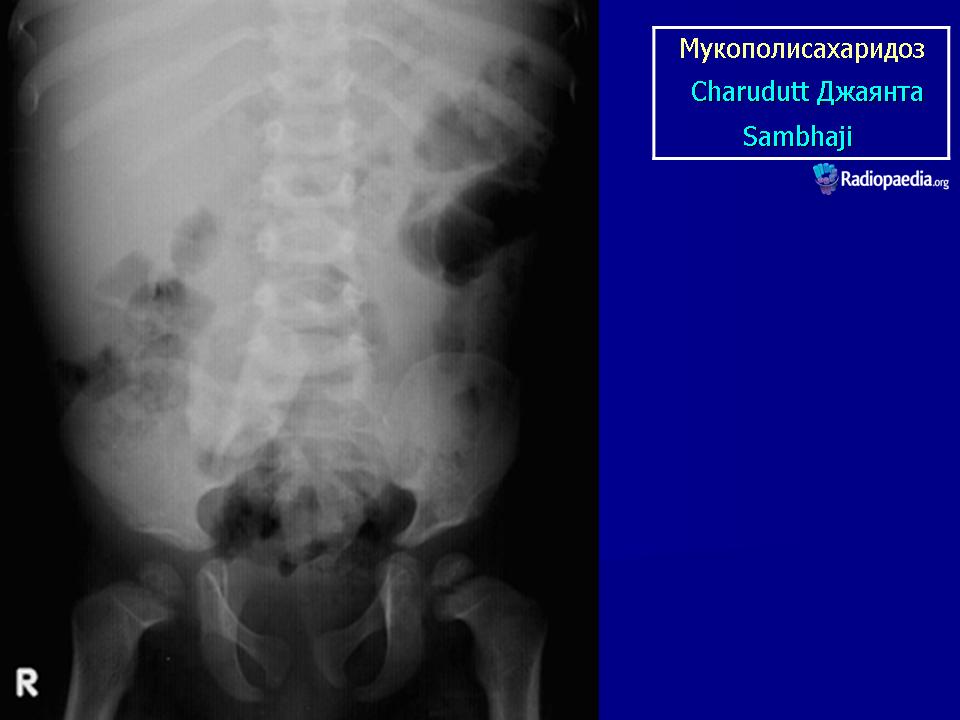
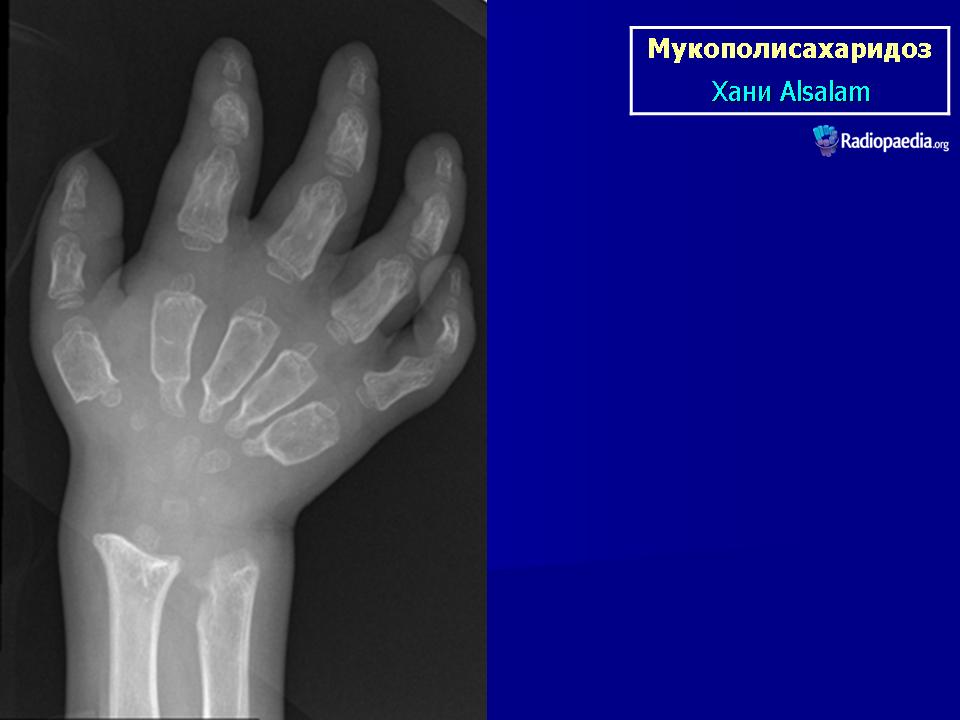
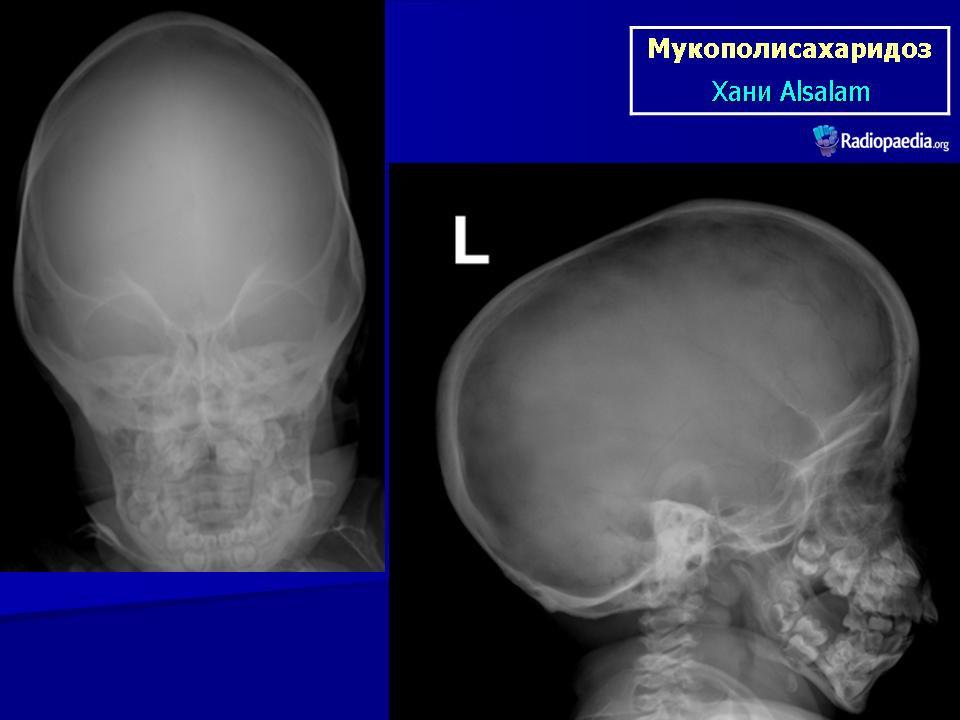
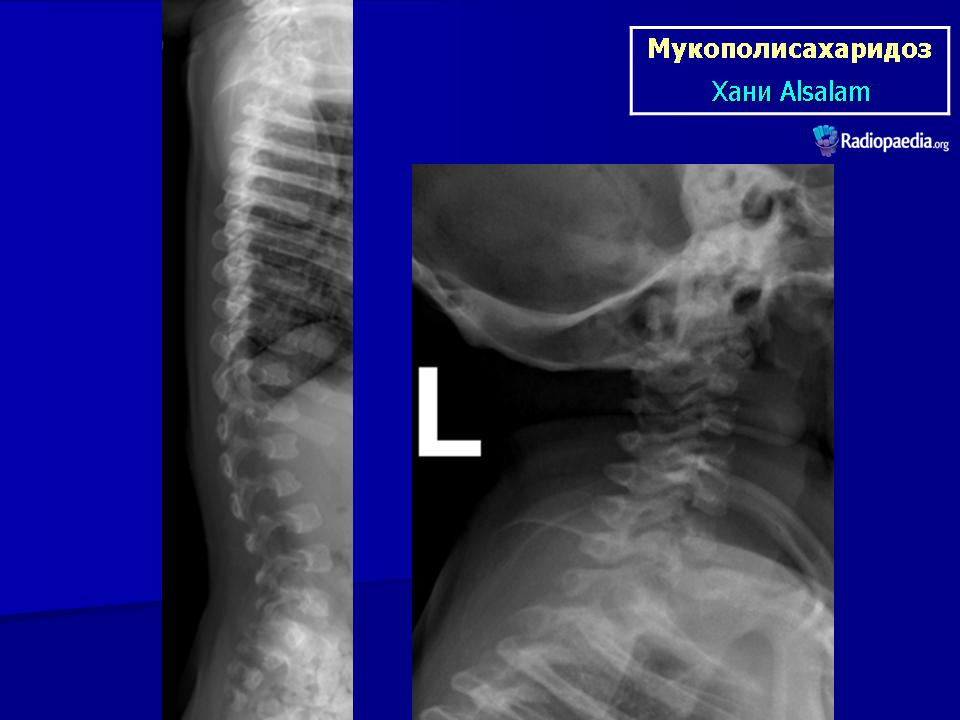
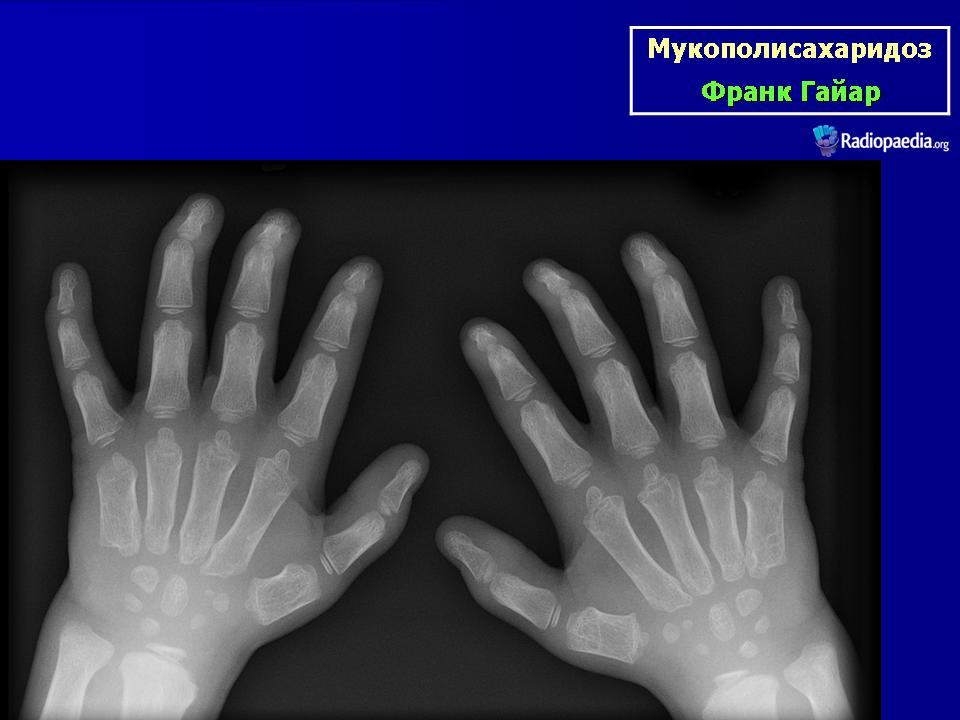
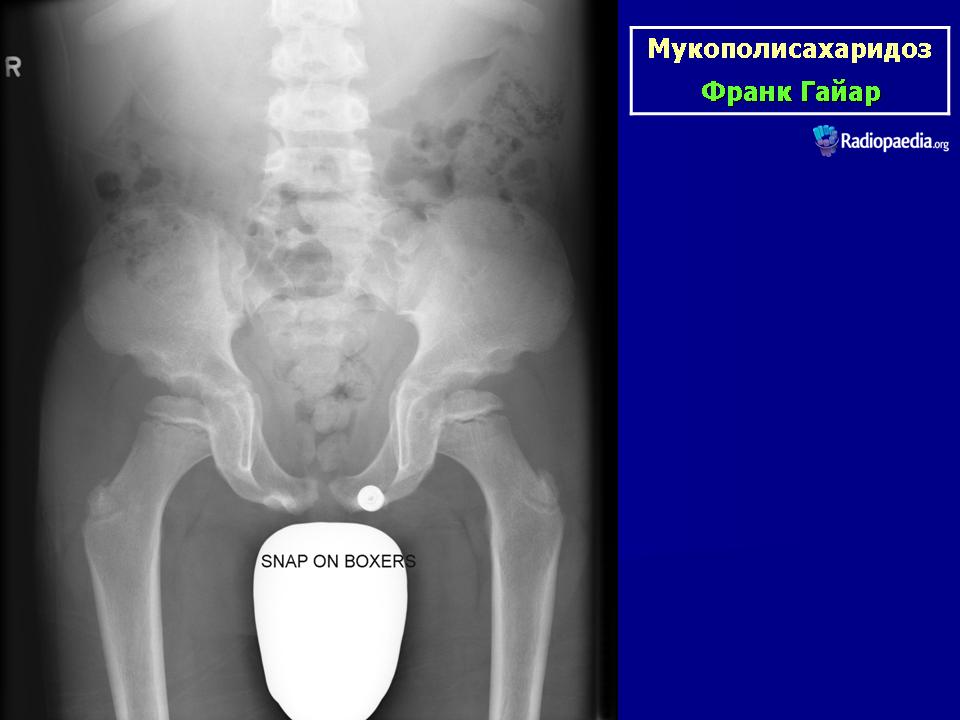
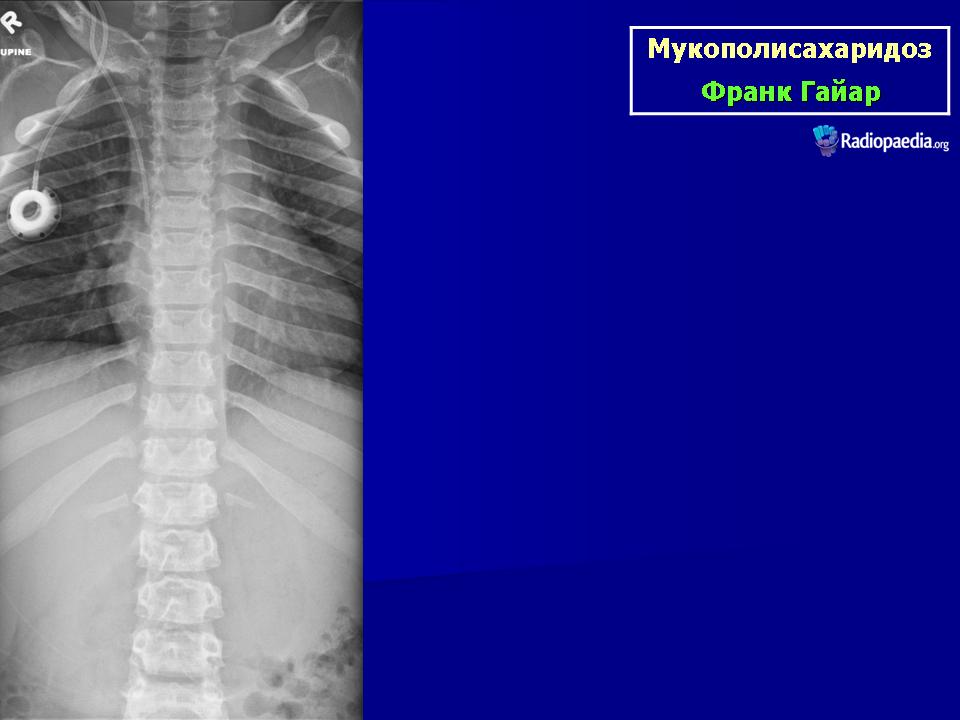
Клиническая симптоматика проявляется уже с рождения характерным внешним видом ребенка — типичные изменения черепа, гротескные черты лица, тугоподвижность многих суставов, абдоминальные грыжи. Заболевание прогрессирует, появляются гепатоспленомегалия, сердечно-сосудистые нарушения, дыхательные расстройства. Часты респираторные инфекции. Характерны также помутнение роговицы, резкое снижение интеллекта и карликовый рост. Течение заболевания злокачественное, приводит к инвалидизации больных в течение нескольких лет.
Мукополисахаридоз II типа (синдром Гунтера) описан S. Hunter в 1919 г. Рецессивный патологический ген локализован в Х-хромосоме, поэтому заболевание проявляется только у мальчиков. Редкие случаи заболевания у девочек, вероятно, являются следствием гомозиготности, возникающей в результате неомутации в Х-хромосоме, полученной от отца.
Синдром обусловлен дефицитом фермента L-идуроно-сульфатсульфатазы, отщепляющей неорганический сульфат от идуроновой кислоты. Ферментативный блок приводит к отсутствию гидролиза дерматан- и гепарансульфата, которые в избыточном количестве, но в меньшем, чем при синдроме Гурлера, экскретируются с мочой.
Заболевание проявляется с рождения, но клинические и патоморфологические нарушения менее выражены, чем при I типе мукополисахаридоза. Деформации черепа и конечностей не столь резкие. Помутнения роговицы не бывает. Интеллект снижен в гораздо меньшей степени. Характерны гепатоспленомегалия, сердечно-сосудистые расстройства. Прогрессируют глухота и снижение зрения. Больные обычно шумливые, несколько агрессивные. Патогномоничными для этой формы мукополисахаридоза считаются множественные участки гладкой, блестящей, непокрытой волосами кожи в области лопаток на фоне общего гирсутизма и утолщения кожи. Течение заболевания медленно прогрессирующее, средняя продолжительность жизни больных составляет около 30 лет.
Мукополисахаридоз V типа (синдром Шейе) описан II. Scheie в 1962 г. и A. Emerit в 1966 г. Наследуется аутосомно-рецессивно. Этот синдром обусловлен мутацией гена, гетероаллельного гену, лежащему в основе мукополисахаридоза I типа Гурлера. Поэтому в некоторых классификациях синдром Шейе называется не мукополисахаридозом V типа, а мукополисахаридозом I типа Шейе. В основе заболевания лежит дефицит фермента a-L-гиалуронидазы, который, однако, менее выражен, чем при синдроме Гурлера.
Характерны тугоподвижность крупных и мелких суставов, деформация стоп, вальгусное искривление коленных суставов. Эти симптомы выражены уже у новорожденных. Дети обычно растут нормально. Интеллект не страдает. Больные имеют специфическое лицо с большим ртом, кожные проявления характеризуются гирсутизмом и утолщением кожи на пальцах.
Мукополисахаридоз VI типа (синдром Марото—Лами, поли дистрофический нанизм). Описан P. Maroteaux и М. Lamy в 1965 г. Обусловлен рецессивным геном, локализованным в аутосоме. В основе патогенеза заболевания лежит дефицит фермента N-ацетилгексозамин-4-SO4-сульфатазы (арилсульфатазы В).
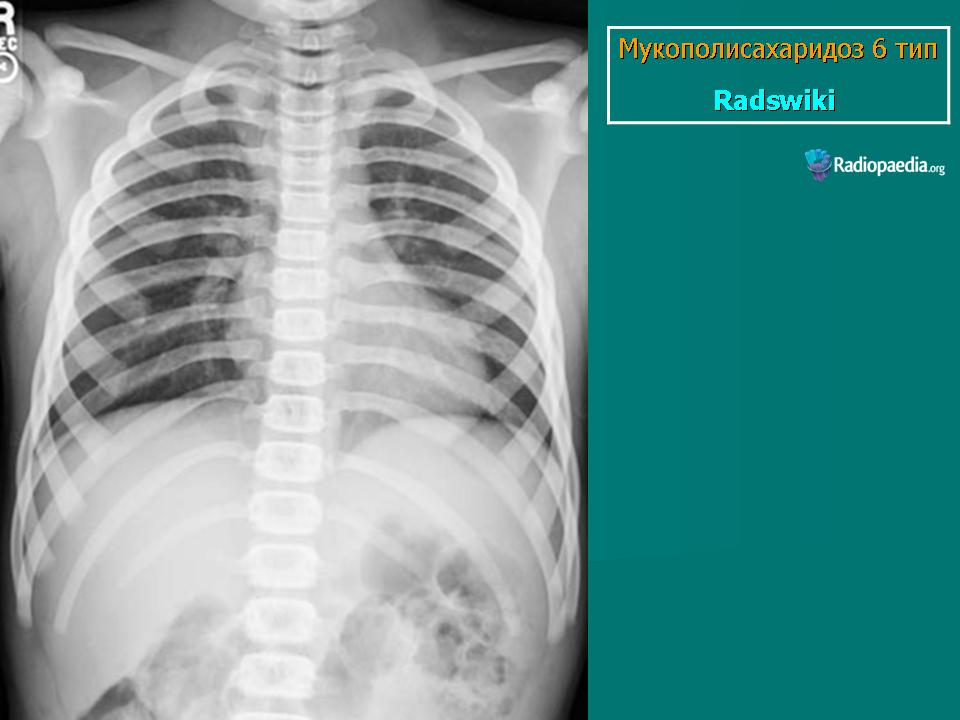
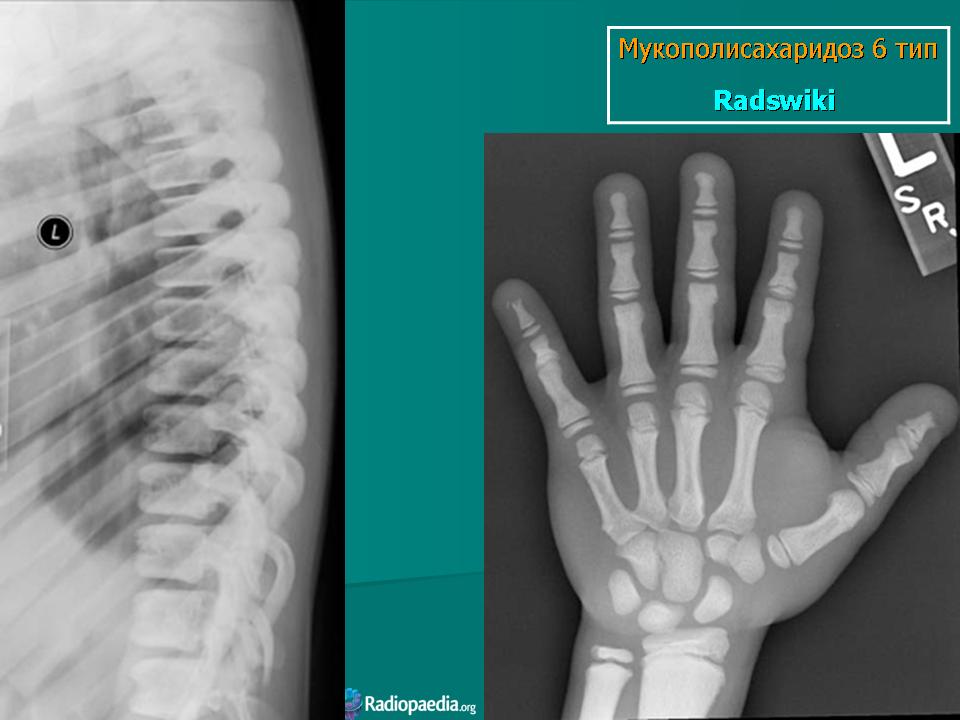
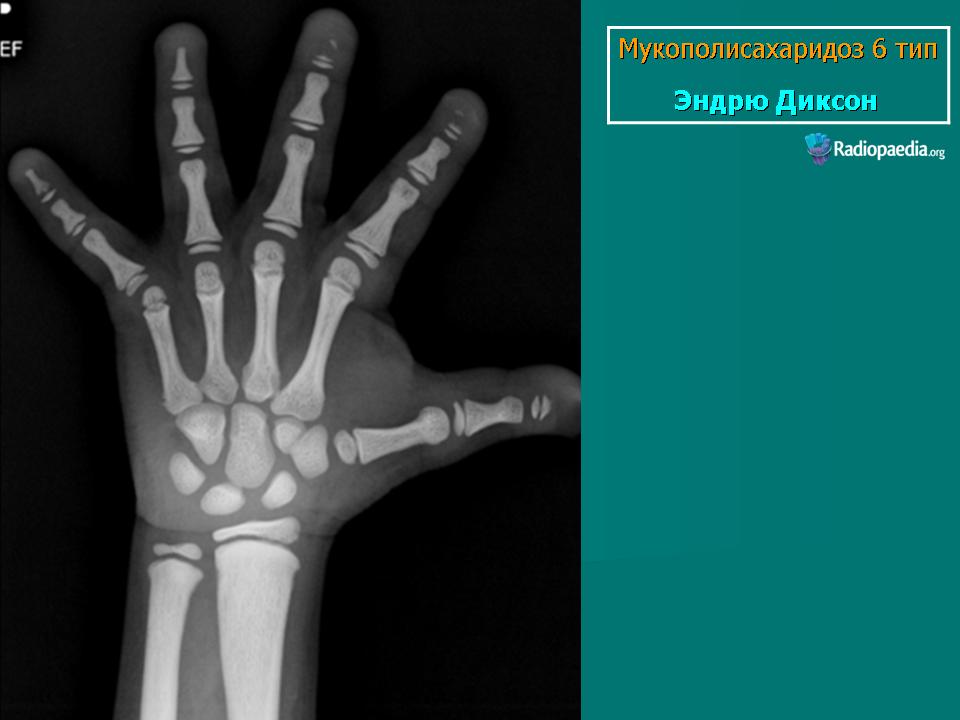
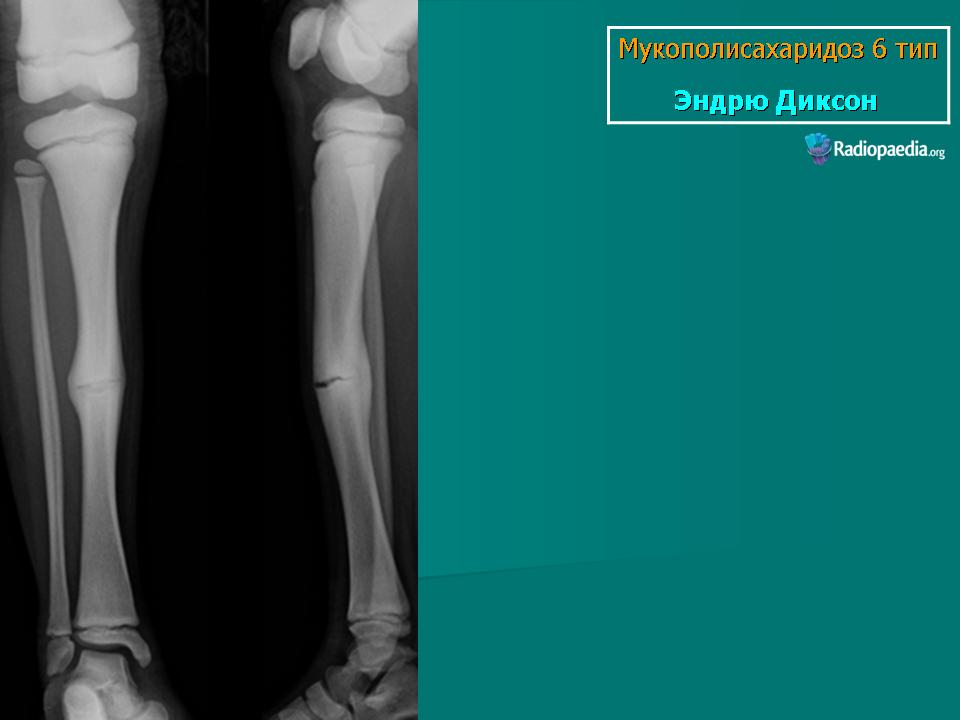
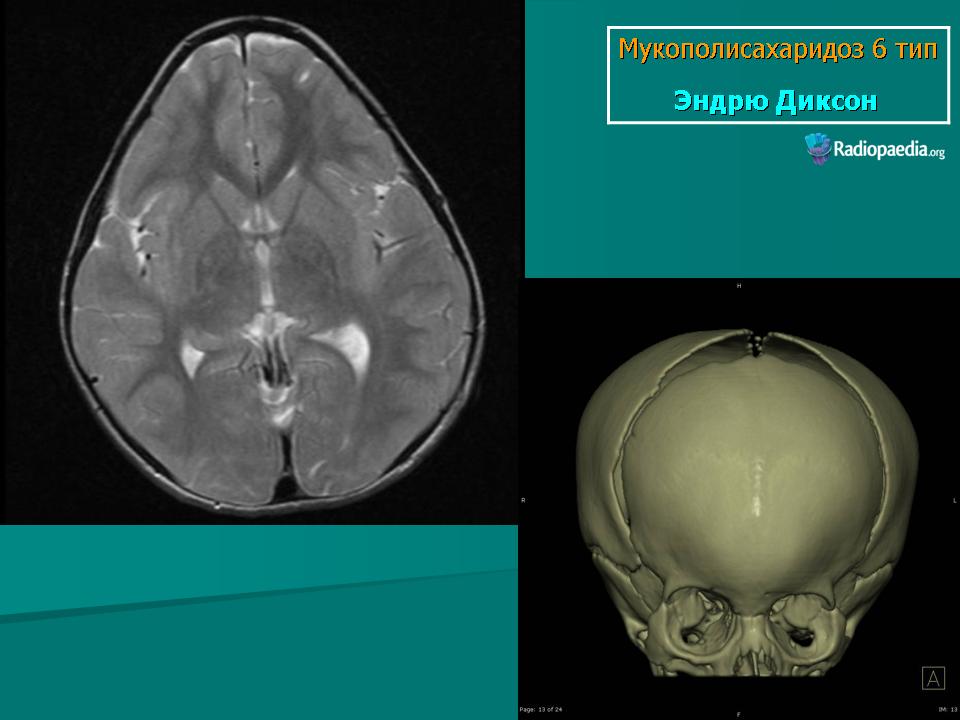
Заболевание проявляется с рождения. Скелетные аномалии и поражение глаз выражены в той же степени, что при синдроме Гурлера. Может развиваться гидроцефалия. Однако психическое развитие страдает незначительно. С мочой экскретируется исключительно дерматансульфат. Мукополисахаридоз VII типа. Описан Н. Sly и др. в 1973 г. Наследуется аутосомно-рецессивно.
При этой форме нарушен катаболизм всех трех фракций гликозамингликанов вследствие дефицита фермента глюкуронидазы. Характерная гротескность черт лица наблюдается уже при рождении. Дети отстают в росте, увеличиваются печень и селезенка. Наблюдаются повторные легочные инфекции. Интеллект сохранен или снижен умеренно. В моче определяется мукополисахаридурия за счет всех трех функций мукополисахаридов. В периферической крови обнаруживаются необычно гранулированные гранулоциты. Течение заболевания медленно прогрессирующее.


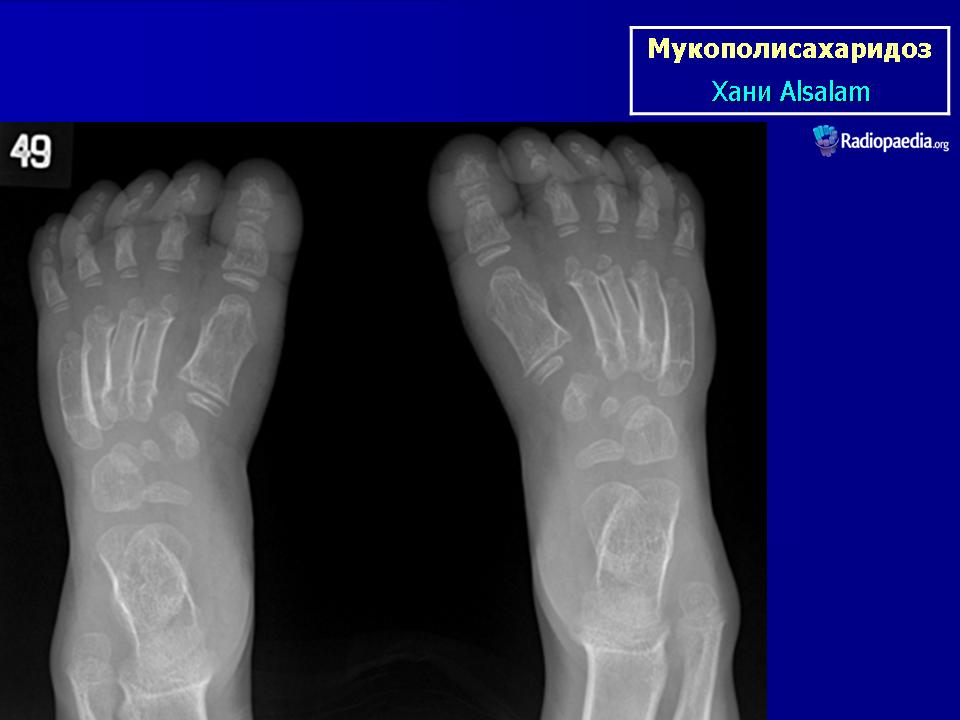

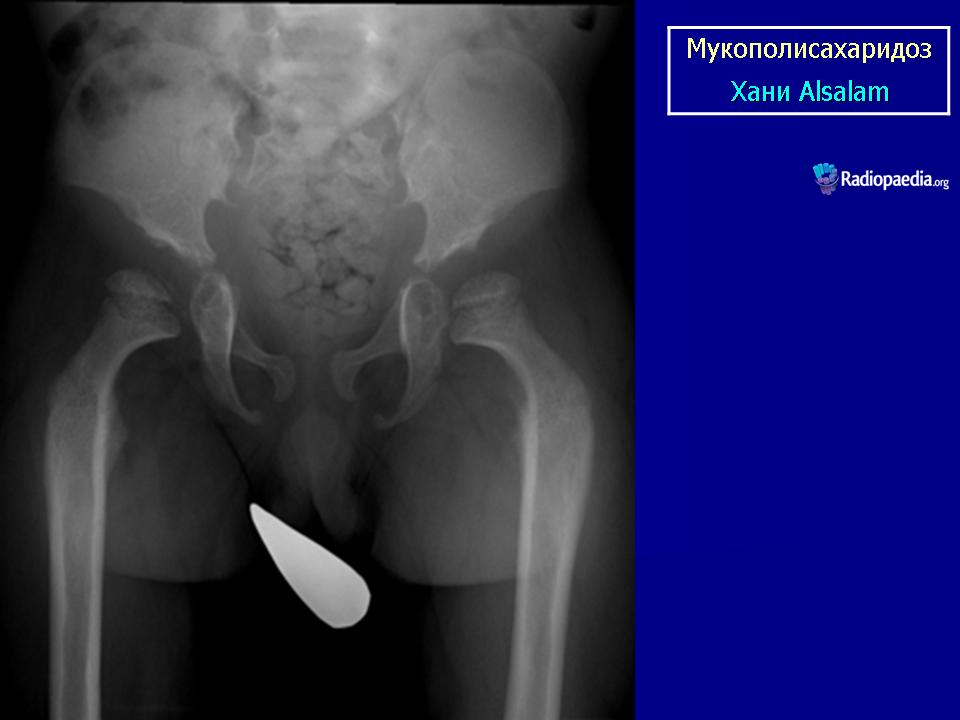
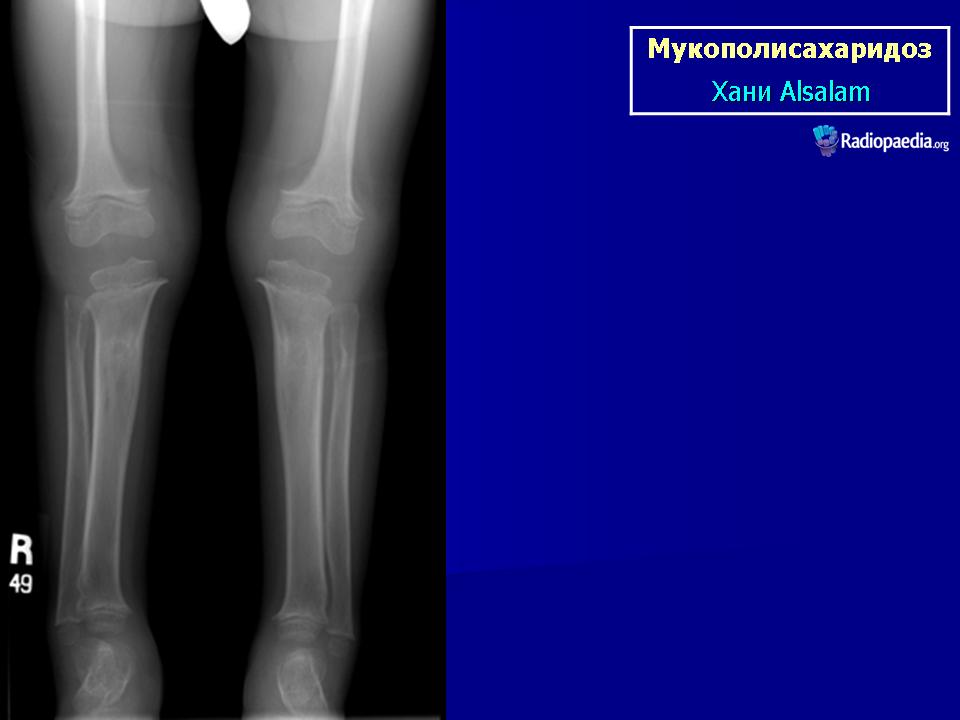
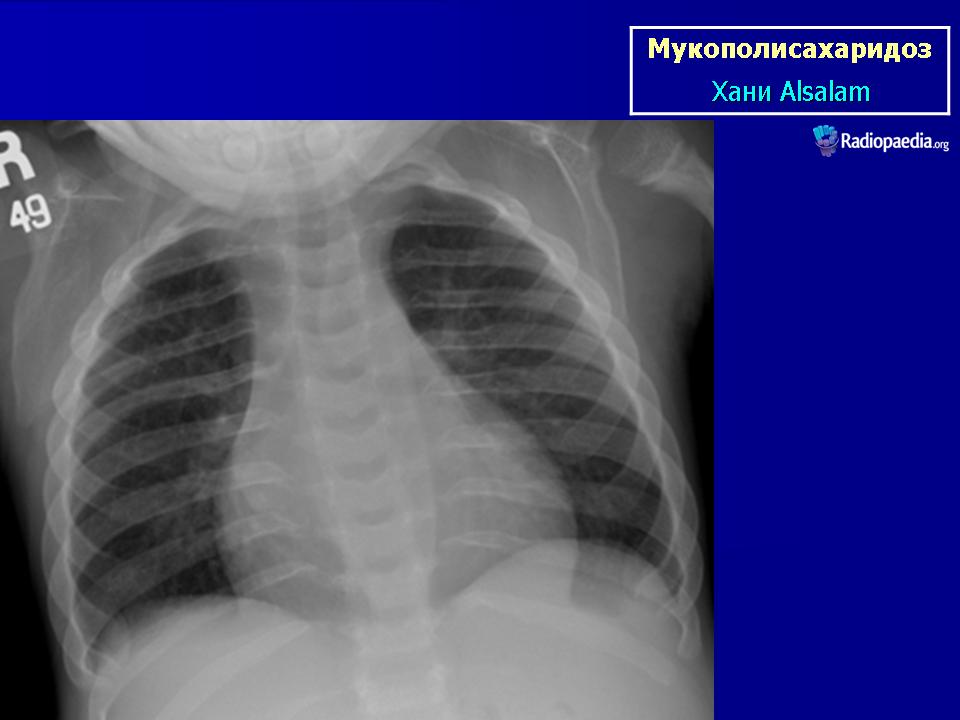




Мукополисахаридоз I типа (синдром Гурлер) описан G. Hurler в 1919 г. Наследуется аутосомно-рецессивно.
В основе патогенеза лежит дефицит фермента а — L-идуронидазы, который приводит к нарушению катаболизма дерматансульфата и гепарансульфата, накапливающихся в клетках и экскретируемых с мочой.
При патоморфологическом исследовании выявляется отложение мукополисахаридов в виде зернистой массы в паренхиме печени, ретикулярных клетках селезенки, эпителии почечных канальцев; сетчатке, склере, роговице глаз; нервных клетках, периферических ганглиях, в интиме коронарных артерий, бронхиальных и других хрящах. Отмечается внутренняя гидроцефалия, обусловленная, по-видимому, отложением мукополисахаридов в мозговых оболочках и нарушением их проницаемости. Определяются очаги демиелинизации. Реактивный фиброз эндокарда связан с отложением мукополисахаридов в клапанах.
Клиническая симптоматика проявляется уже с рождения характерным внешним видом ребенка — типичные изменения черепа, гротескные черты лица, тугоподвижность многих суставов, абдоминальные грыжи. Заболевание прогрессирует, появляются гепатоспленомегалия, сердечно-сосудистые нарушения, дыхательные расстройства. Часты респираторные инфекции. Характерны также помутнение роговицы, резкое снижение интеллекта и карликовый рост. Течение заболевания злокачественное, приводит к инвалидизации больных в течение нескольких лет.
Мукополисахаридоз II типа (синдром Гунтера) описан S. Hunter в 1919 г. Рецессивный патологический ген локализован в Х-хромосоме, поэтому заболевание проявляется только у мальчиков. Редкие случаи заболевания у девочек, вероятно, являются следствием гомозиготности, возникающей в результате неомутации в Х-хромосоме, полученной от отца.
Синдром обусловлен дефицитом фермента L-идуроно-сульфатсульфатазы, отщепляющей неорганический сульфат от идуроновой кислоты. Ферментативный блок приводит к отсутствию гидролиза дерматан- и гепарансульфата, которые в избыточном количестве, но в меньшем, чем при синдроме Гурлера, экскретируются с мочой.
Заболевание проявляется с рождения, но клинические и патоморфологические нарушения менее выражены, чем при I типе мукополисахаридоза. Деформации черепа и конечностей не столь резкие. Помутнения роговицы не бывает. Интеллект снижен в гораздо меньшей степени. Характерны гепатоспленомегалия, сердечно-сосудистые расстройства. Прогрессируют глухота и снижение зрения. Больные обычно шумливые, несколько агрессивные. Патогномоничными для этой формы мукополисахаридоза считаются множественные участки гладкой, блестящей, непокрытой волосами кожи в области лопаток на фоне общего гирсутизма и утолщения кожи.
Течение заболевания медленно прогрессирующее, средняя продолжительность жизни больных составляет около 30 лет.
Мукополисахаридоз V типа (синдром Шейе) описан II. Scheie в 1962 г. и A. Emerit в 1966 г. Наследуется аутосомно-рецессивно. Этот синдром обусловлен мутацией гена, гетероаллельного гену, лежащему в основе мукополисахаридоза I типа Гурлера. Поэтому в некоторых классификациях синдром Шейе называется не мукополисахаридозом V типа, а мукополисахаридозом I типа Шейе.
В основе заболевания лежит дефицит фермента a-L-гиалуронидазы, который, однако, менее выражен, чем при синдроме Гурлера.
Характерны тугоподвижность крупных и мелких суставов, деформация стоп, вальгусное искривление коленных суставов. Эти симптомы выражены уже у новорожденных. Дети обычно растут нормально. Интеллект не страдает. Больные имеют специфическое лицо с большим ртом, кожные проявления характеризуются гирсутизмом и утолщением кожи на пальцах.
Мукополисахаридоз VI типа (синдром Марото—Лами, поли дистрофический нанизм). Описан P. Maroteaux и М. Lamy в 1965 г. Обусловлен рецессивным геном, локализованным в аутосоме.
В основе патогенеза заболевания лежит дефицит фермента N-ацетилгексозамин-4-SO4-сульфатазы (арилсульфатазы В).
Заболевание проявляется с рождения. Скелетные аномалии и поражение глаз выражены в той же степени, что при синдроме Гурлера. Может развиваться гидроцефалия. Однако психическое развитие страдает незначительно. С мочой экскретируется исключительно дерматансульфат.
Мукополисахаридоз VII типа. Описан Н. Sly и др. в 1973 г. Наследуется аутосомно-рецессивно.
При этой форме нарушен катаболизм всех трех фракций гликозамингликанов вследствие дефицита фермента глюкуронидазы.
Характерная гротескность черт лица наблюдается уже при рождении. Дети отстают в росте, увеличиваются печень и селезенка. Наблюдаются повторные легочные инфекции. Интеллект сохранен или снижен умеренно. В моче определяется мукополисахаридурия за счет всех трех функций мукополисахаридов.
В периферической крови обнаруживаются необычно гранулированные гранулоциты. Течение заболевания медленно прогрессирующее.
Продолжение.
Мукополисахаридоз
Из архива AFIP
Продолжение.
Мукополисахаридоз У1 типа.
https://radiopaedia.org/articles/pyknodysostosis
Дифференциальная диагностика
https://radiopaedia.org/articles/mucopolysaccharidoses-2