Стерджа (Штурге) (Sturge) - Вебера (Weber) синдром (болезнь)
Синдром, описанный в 1879 году Sturge , дополненный позже Weber (1922) и Krabbe (1934), представляет собой полную форму энцефало-лицевого нейроангиоматоза, характеризующуюся сочета-ниемкожного и мозгового ангиоматоза с глазными проявлениями. Синдром известен и множеством синонимов: "синдром Weber ", "синдром Sturge - Weber - Dimitri ", "синдром Weber - Dimitri ", "синдром Sturge ", "синдром Kallischer ", "синдром Krabbe ", "синдром Sturge - We - ber - Krabbe - Brushfield - Wyatt ", "синдром Lawford ", "синдром Schirmer ", "синдром Milles ", "врожденная нейроэктодермальная дисплазия", "врожденный эктодермоз", "кожно-мозговая ангиома", "энцефало-лицевой нейроангиоматоз", "энцефалотройничный ангиоматоз", "ангиоматоз мозговых оболочек, глаз и лица".
Симптоматология
Кожный ангиоматоз. Присутствует от рождения или развивается в первом детстве, в форме ангиом лица, названных ,, naevus flammeus vasculosus "; эти пятна тёмнокрасного цвета, плоские (не выступающие на поверхности покровов) локализованы чаще всего на одной половине лица, на территории тройничного нерва. Часто ,, naeveus flammens " намного переходит границы территории тройничного нерва, захватывали часть шеи, грудной клетки и даже живота; редко появляется и на слизистой десен, рта, губ и носа. Одновременно с ,, naevm flammeus " на лице могут появиться и небольшие телеангиэктические пятна.
Кожный ангиоматоз является следствием расширения капилляров, обусловленного врожденным отсутствием резистентности сосудистой стенки.
Односторонний ангиоматоз могза. В мозговом веществе и на его оболочках появляются венозные бородавки и телеангиэктазии. Часто наблюдается неравномерность развития и нарушения черепно-мозговой симметричности, половина черепа, пораженная ангио-матозным процессом будучи меньшей, чем противоположная.
Клинически ангиоматоз мозга проявляется:
-
эпилептическими припадками, в начале половины тела, а затем общими и появляющимися в очень раннем возрасте;
-
приступами мигрени (только у большого ребенка);
-
афазией;
-
гемипарезами: моно- или гемиплегамиями;
-
олигофренией или сумашествием (приблизительно в 60% случаев).
Иногда встречаются изменения костей лица (гипертрофия лицевого массива и свода черепа).
Глазные признаки постоянные и тяжелые:
-
ангиома хороидеи, всегда распложенная в сосочко-макулляр-ной области. Ангиома имеет вид серовато-белого пятна, в форме диска; со временем может прогрессивно развиваться или ее рост прекращается, и она кальцифицируется;
-
врожденная глаукома, со стороныангиоматозныхпоражений кожи, мозга, глаза; гидрофтальмия со стороны ангиоматоза; гемиа-нопсия; ангиома конъюнктивы.
Помимо полной клинической формы, существуют и формы атипические, с бедной симптоматологией. Некоторые из них были индивидуализированы как следующие синдромы:
-
Jahnke (1930), при котором отсутствует глаукома;
-
Schirmer (1860), при котором глаукома и гидрофтальмия появляются рано;
-
Lawford (1884), при котором глаукома появляется поздно, имеет хроническое течение и не вызывает увеличение объема глазного яблока;
- Milles (1884): существует ангиома хороида, но без увеличения объема глазного яблока.
Снимки черепа выявляют наличие некоторых внутричерепных кальцификаций, имеющих вид "мозговидных штрихов", похожих на извилистые линии, наслоенные на извилины мозга, чаще в затылочной доле; подобный аспект встречается очень часто у большого ребенка и только редко у грудного.
Ангиография мозга может указать на существование и расположение мозговой ангиомы.
Газовая энцефалография и электроэнцефалограмма выявляют косвенные признаки атрофии или страдания мозга. Часто отмечается уменьшение биоэлектрической активности в кальцифицированных зонах мозга.
11 атологоанатомическое обследование доказывает, что ангиоматоз заинтересовывает венозную систему, часто и каплиляры. На мягкой оболочке мозга неравномерные сосудистые расширения, распространенные всегда односторонне и обычно в затылочной области. Нижележащие извилины атрофичны, а находящиеся вокруг них клетки коры мозга - дегенеративны. В коре находятся отложения кальция в стенках мелких сосудов. В глазах отмечаются ангиомы хороидеи.
Этиопатогенез. Этиология неизвестна. Предполагается существование порока мезодермы, в которой зарождается мозговая сосудистая система; следовательно, речь идет о врожденной дисплазии. Наследственность синдрома явно еще не доказана, но несколько наблюдений показывают неравномерную доминурующую передачу. Поражаются оба пола. В большинстве опубликованных случаев, возраст больных колеблется между 10-20 годами, что доказывает повышенную частоту синдрома среди больших детей и среди молодых взрослых.
Эволюция и прогноз: зависят от распространенности мозговых поражений. Обычно прогноз неблагоприятный из-за частых и тяжелых нейрологических осложнений.
Лечение. Этиологического лечения не существует.
Хирургическое лечение состоит в рассечении доли или в удалении половины мозга, с целью радикального устранения ангиомы мозга; операция показана в первых стадиях болезни, когда нейрологические и психические проявления еще не запущены. Хирургическое лечение показано иногда и для корригирования глазных поражений.
При кожных ангиомах можно получить хорошие результаты при помощи рентгенотерапии.
Вторичная эпилепсия у ребенка, в особенности после 2-летнего возраста, вообще устойчива к противососудным препаратам и требуется много терапевтических попыток для нахождения самого эффективного противоэпилептического вещества.

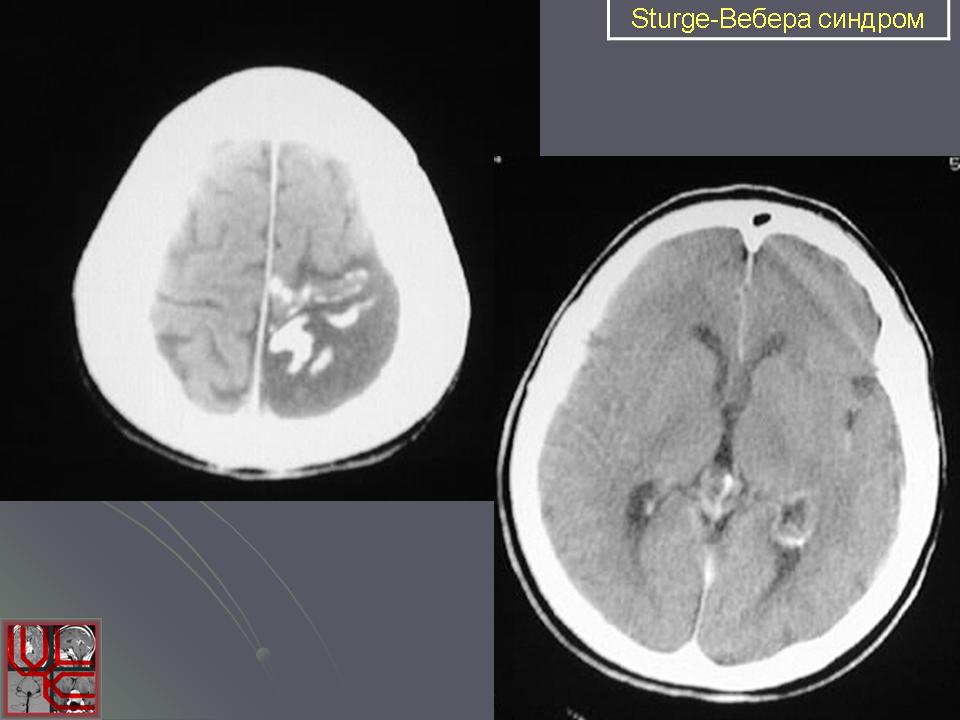
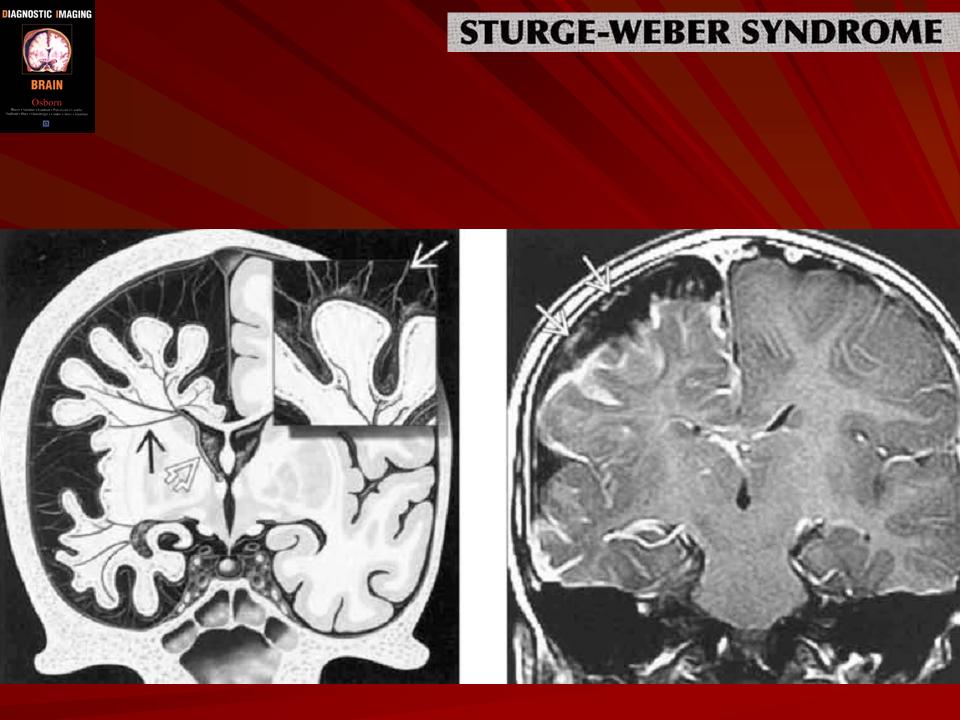

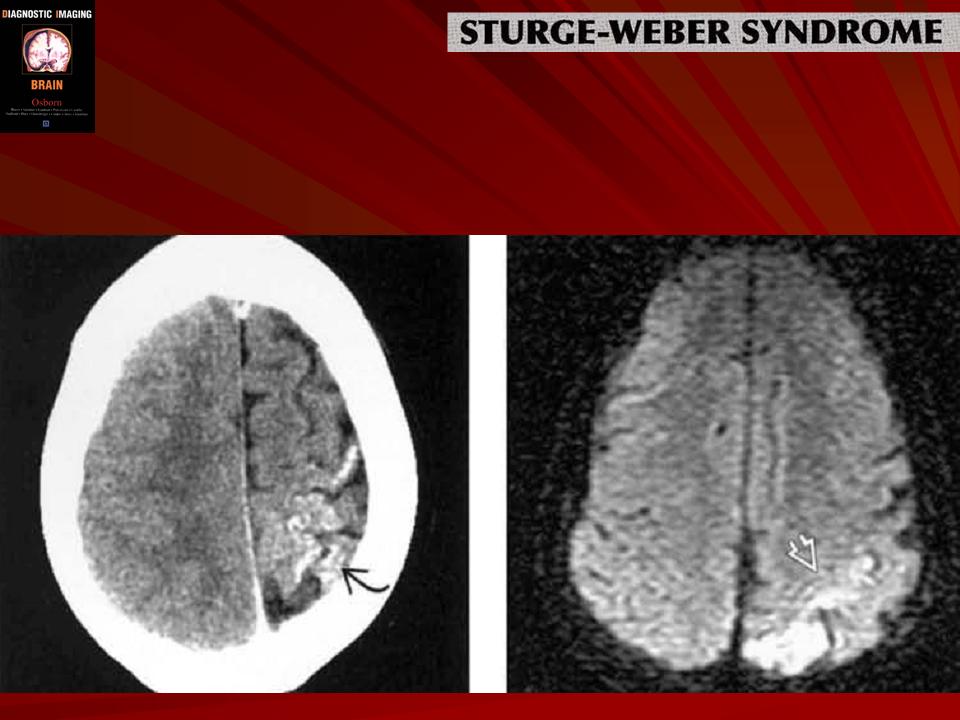

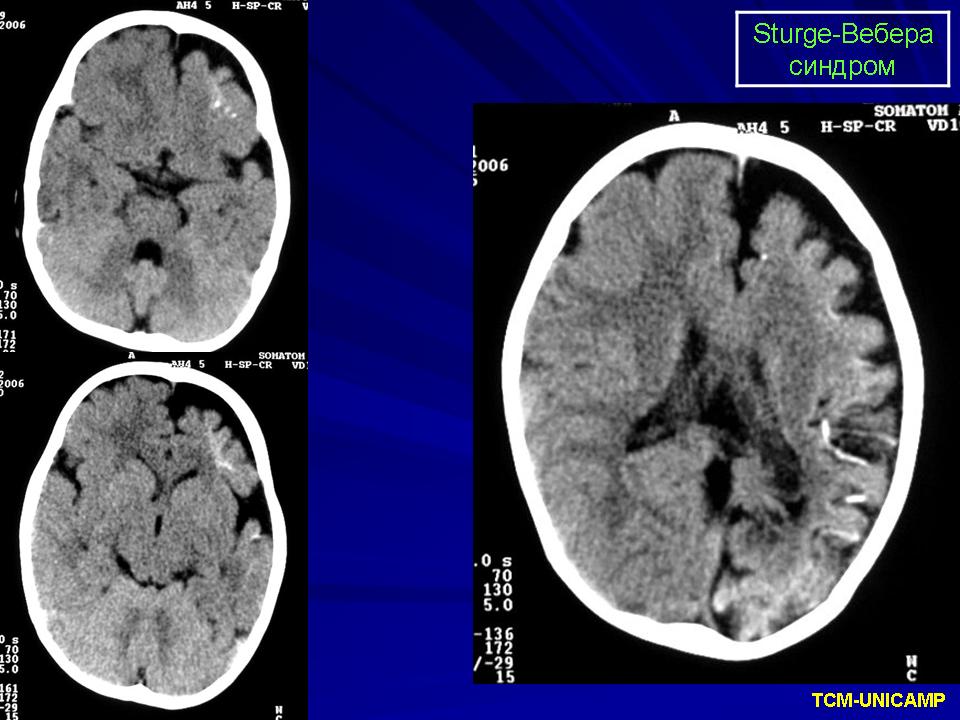
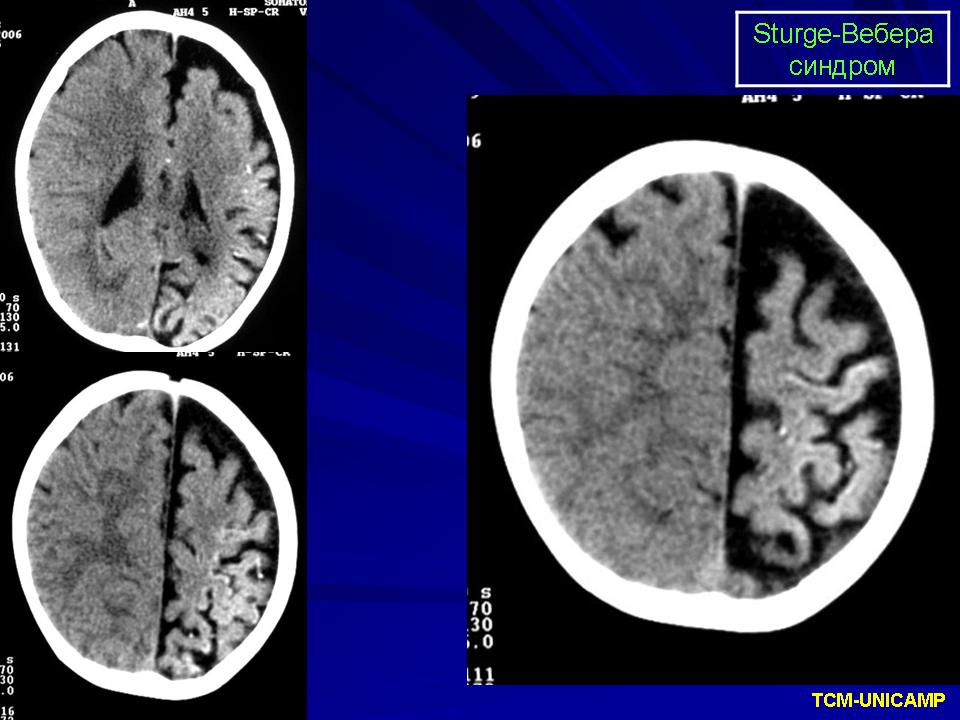
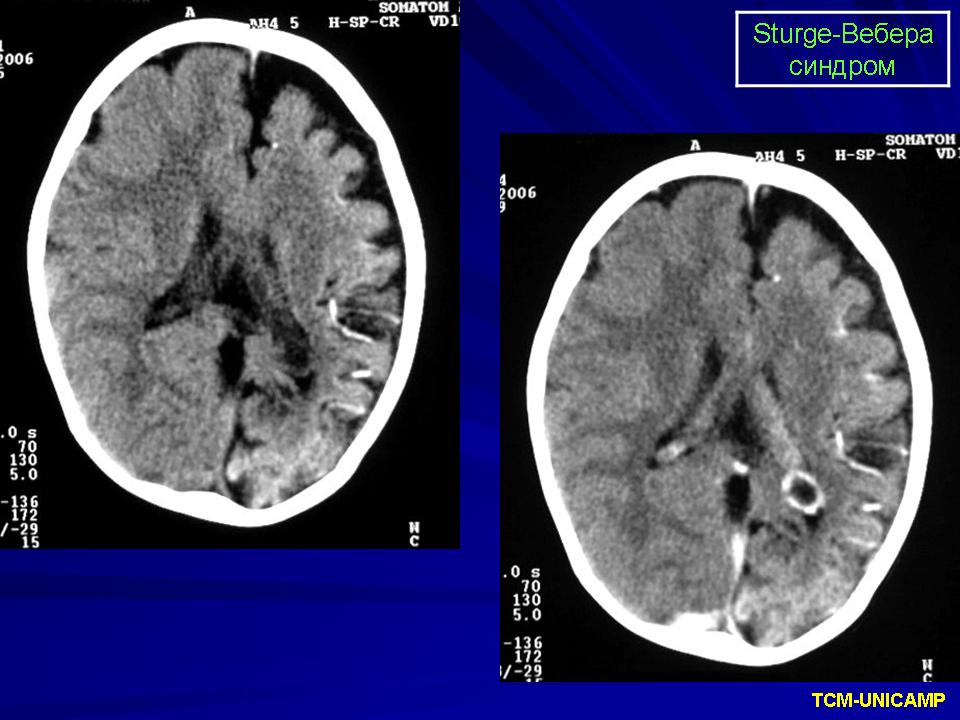
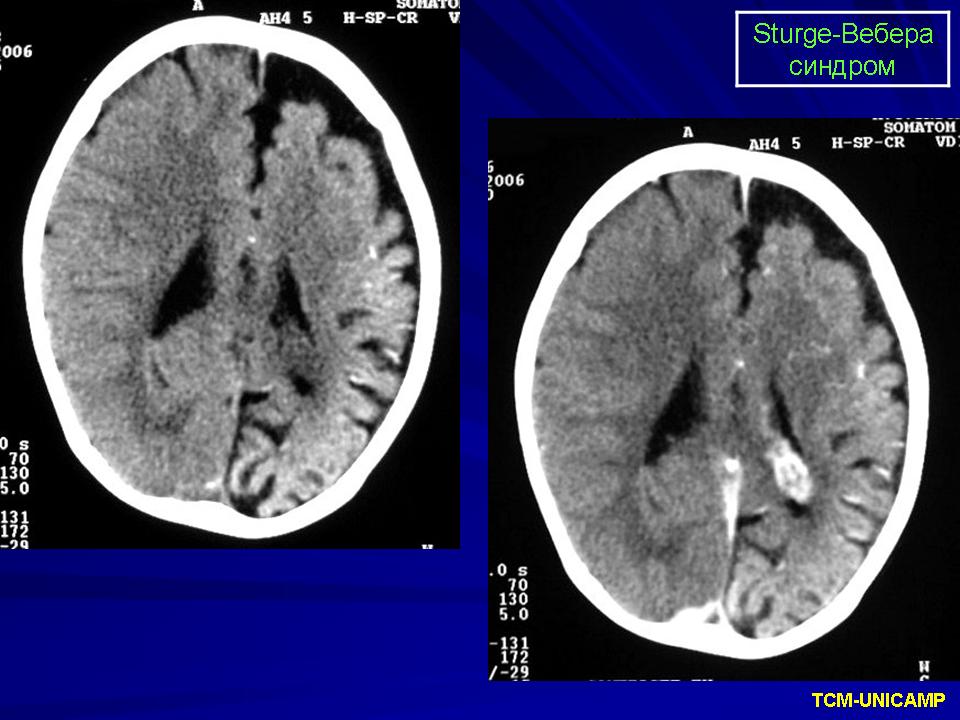
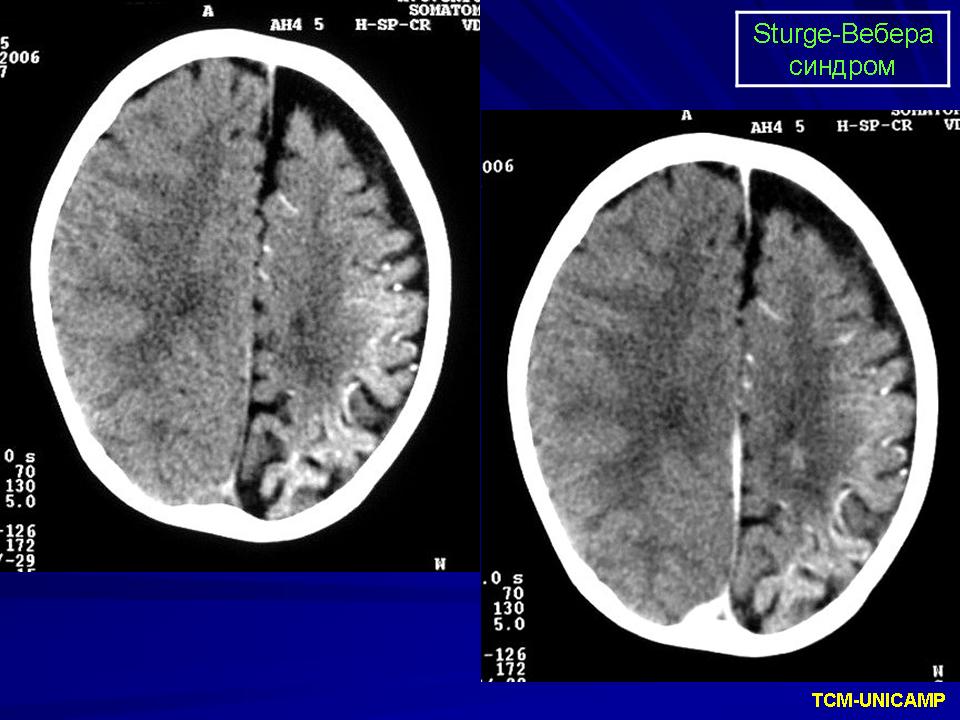
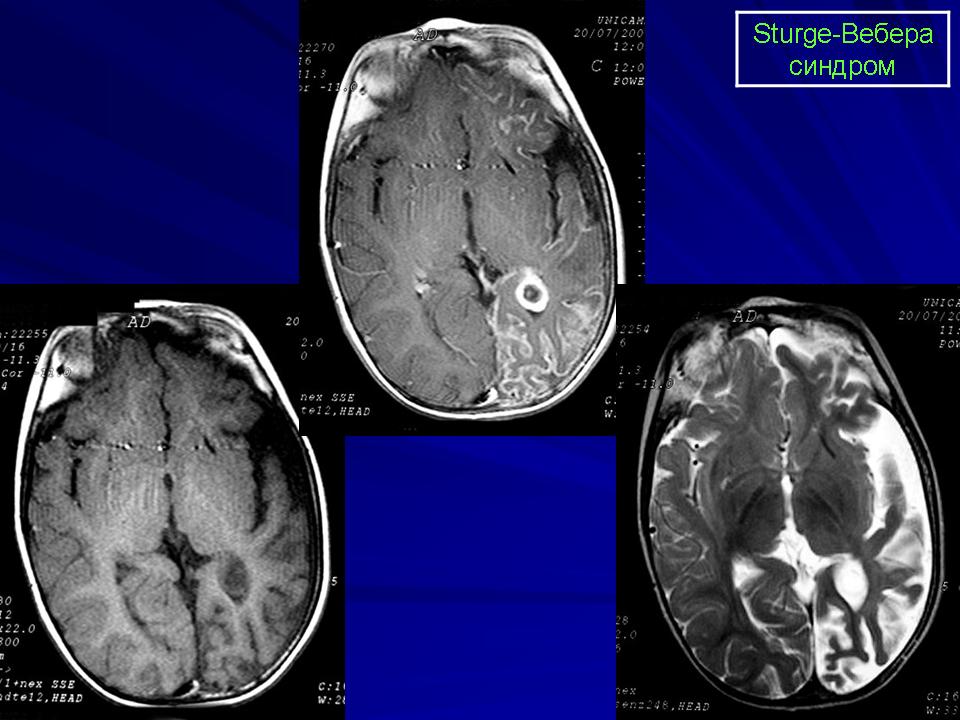
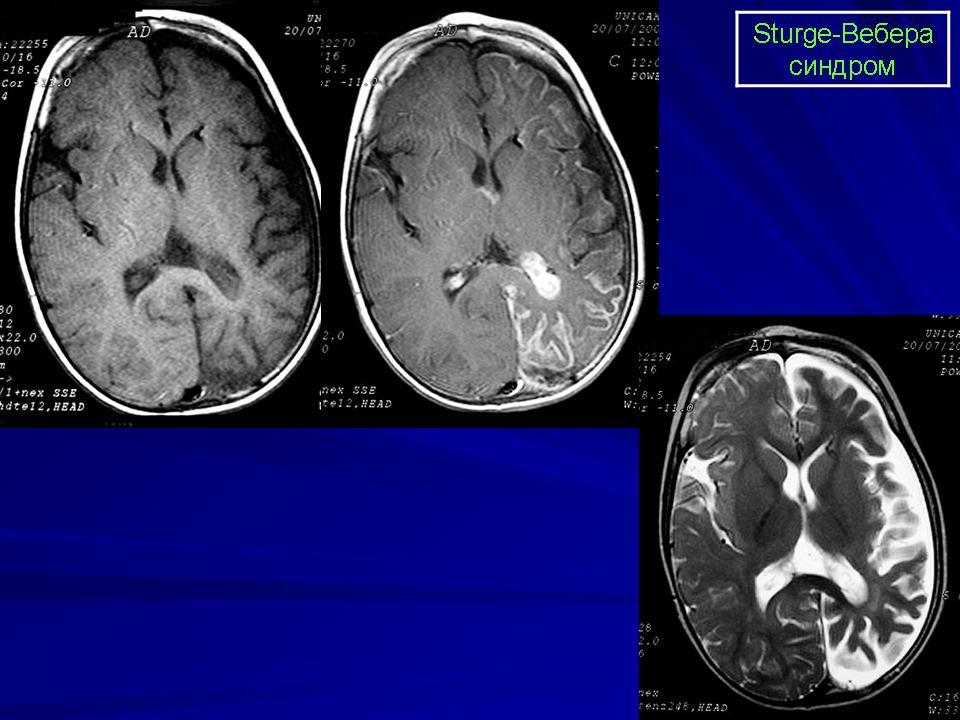
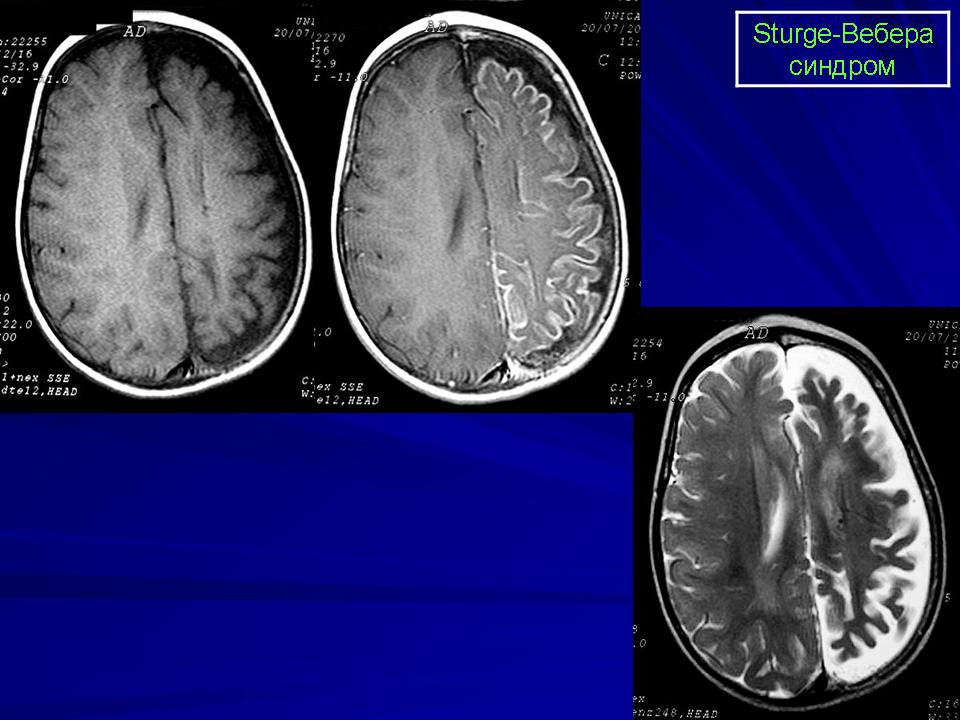

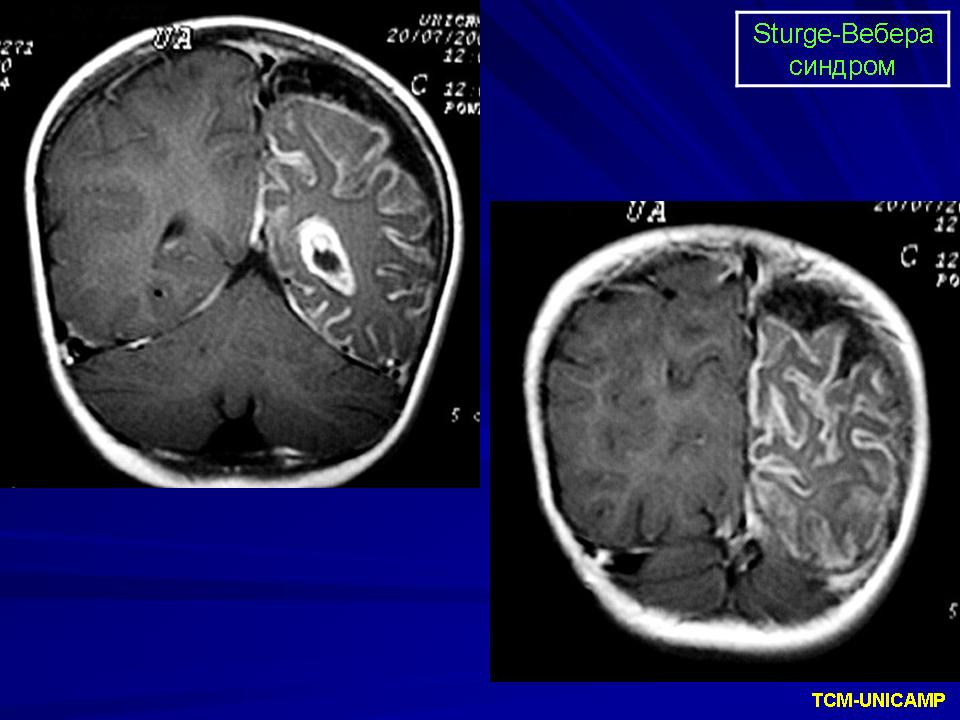
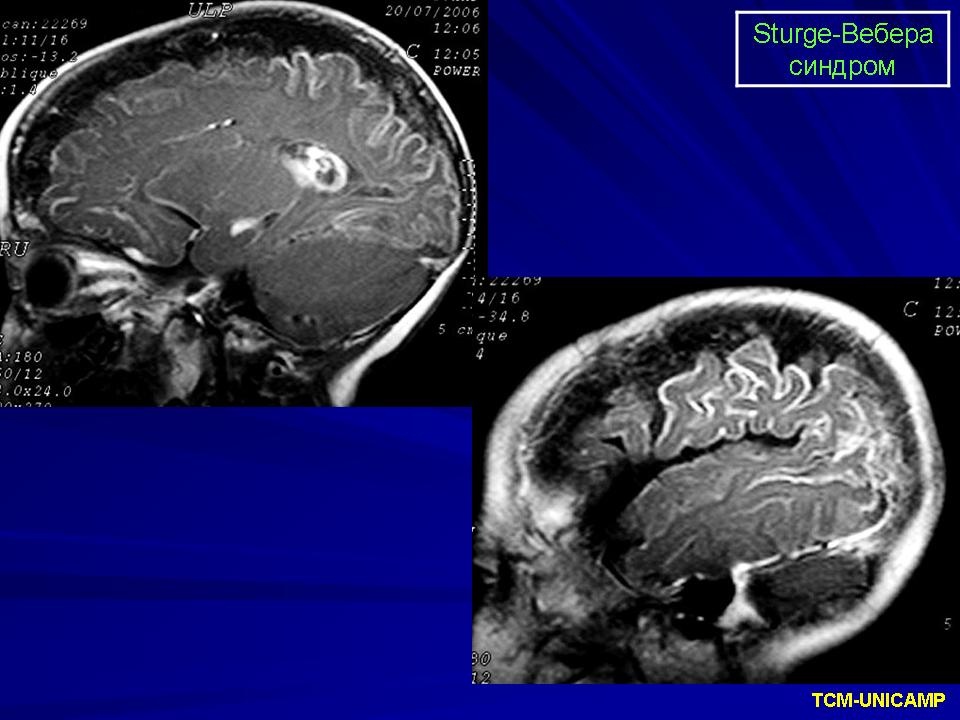
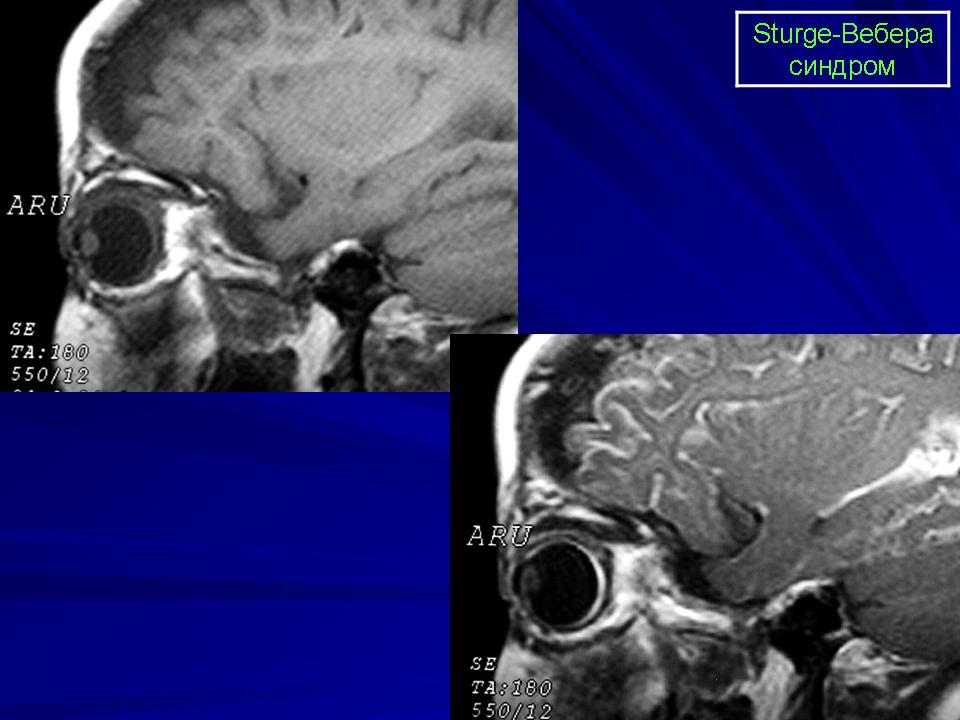

Синдром Sturge-Weber.
синдром Weber, синдром Sturge-Weber-Dimitri, tnorbus Sturge, синдром Kalischer, синдром Krabbe, синдром Brushfield-Wyatt, neuroangiomatosis encephalofacialis, angioma capillare et venosum calcificans, ectodermosis congenita.
Sturge William Allen (1850—1919); Weber Frederick Parkes (1863—1962), английские врачи.
Стерджа — Вебера с. — врожденный ангиоматоз в области тройничного нерва и нарушение развития мозга: обычно односторонние naevus flammeus в области иннервации тройничного нерва, врожденная (гомо-или билатеральная) глаукома; эпилептиформные припадки; часто спастические параличи, нередко олигофрения. Рентгенологически — на стороне гемангиомы в костях черепа — дважды контурированные обызвествления. На энцефалограмме часто расширение желудочков. Встречается сочетание с аномалиями других органов. Предполагается также аутосомно-доминантное наследование.
Продолжение.
Синдром Sturge-Weber.
Синдром Sturge-Weber.
Синдром Sturge-Weber.
Заболевание относится к группе факоматозов. Факоматозы представляют собой наследственные заболевания, характеризующиеся изменениями кожных покровов, наиболее частыми из которых являются пятна (phacos - пятно), неврологическими нарушениями и патологией внутренних органов. Такая комбинация связана с нарушением развития одновременно двух зародышевых листков - эктодермы и мезодермы из этих зародышевых зачатков формируются нервная система кожа и ее производные, внутренние органы и сосуды. К этой группе заболеваний относятся энцефалотригеминальный ангиоматоз Штурге-Вебера, туберозный склероз Бурневилля, синдром Луи-Бар (атаксия-телеангиэктазия), нейрофиброматоз Реклингхаузена. Многие формы патологии передаются из поколения в поколение. Общей клинической закономерностью для заболеваний этой группы является неуклонное прогрессирование симптомов, нередко приводящее к тяжелым необратимым изменениям многих функций организма. Классическое проявление болезни характеризуется триадой симптомов: сосудистыми пятнами на коже лица (ангиомы), судорожными приступами (чаще парциальные), повышением внутриглазного давления (глаукома).
Патология часто носит семейный характер с аутосомно-доминантным наследованием, но описаны и аутосомно-рецессивные формы. Ангиомы на коже обычно обнаруживаются уже при рождении, имеют вид «пылающего пятна». В 80% случаев они располагаются на лице (с одной или двух сторон) в области иннервации ветвей тройничного нерва. Судорожные припадки появляются в первые годы жизни, обычно носят очаговый характер. У многих больных они заканчиваются генерализованным судорожным припадком. Возможны бессудорожные приступы в виде мгновенных отключений сознания, вздрагиваний, застываний. У некоторых больных возникают сильные приступы головной боли с рвотой (мигренеподобные приступы). Повышение внутриглазного давления (глаукома) наблюдается с рождения или появляется позднее. Частота появления глаукомы составляет около 50%. Прогрессирование глаукомы приводит к снижению зрения вплоть до полной слепоты. Из других проявлений болезни часто встречается слабоумие, которое обусловлено повторяющимися приступами. Оно сочетается с выраженными изменениями в эмоционально-волевой сфере: злопамятностью, эгоцентризмом, аффективностью, мстительностью. Ухудшаются память, внимание, способность усваивать новые сведения. Указанные факторы значительно осложняют процесс обучения и воспитания. Эти нарушения психики отмечаются и в период между приступами. Выраженность расстройств интеллекта, особенно памяти, нарастает по мере прогрессирования заболевания и учащения судорог. Диагноз энцефалотригеминального ангиоматоза подтверждается при измерении внутриглазного давления, исследовании глазного дна, рентгенографией черепа и записи биотоков мозга.
Магнитная резонансная томография пациента с болезнью Штурге - Вебера
При патоморфологическом исследовании больных, страдавших энцефалотригеминальным ангиоматозом, выявляется разрастание сосудов кожи, мягкой мозговой оболочки, сосудистых сплетений глазного яблока. Реже ангиомы локализуются в затылочной области больших полушарий, мозжечке, спинном мозге и внутренних органах. Описано отложение солей кальция в сосудах мозга (петрификаты). Выраженность ангиом может значительно варьироваться. При лечении заболевания используют противосудорожные и психотропные средства, а также препараты, снижающие внутричерепное и внутриглазное давление.
Показанием к нейрохирургической операции является наличие некупируемых приемом противосудорожных препаратов судорог. В таком случае может быть выполнена резекция части очагов или гемисферэктомия.
Синдром Sturge-Weber.
В 1879 г. Sturge описал пациента, страдавшего гемипарезом и эпилептическими припадками, у которого на лице имелась ангиома. В 1922 г. Weber дополнил клиническую картину описанием обызвествлений на рентгенограммах у больных этим синдромом. В 1955 г. он же обозначил заболевание термином «энцефалотригеминальный ангиоматоз».
Основными клиническим симптомами заболевания являются врожденные сосудистые пятна на лице и эпилептические припадки. Кроме того, могут наблюдаться гемипарезы, гемианопсия, гидрофтальм, глаукома, умственная отсталость. Синдром встречается с частотой 1 на 100 ООО рождений. Заболевание чаще носит спорадический характер, но имеются описания наследственных случаев с доминантным и рецессивным типами наследования и незначительной пенетрантностью.
Патологическая анатомия синдрома Стержа-Вебера
Типичными морфологическими компонентами синдрома являются сосудистые пятна на лице и ангиоматоз оболочек мозга, представляющий собой сеть венул, располагающихся в мягкой оболочке конвекситальной поверхности мозга, чаще в заднетеменных или затылочных его отделах. Пятна и оболочечные ангиомы обычно располагаются на одной стороне. Как правило, в этих же участках отмечается атрофия и обызвествление коры мозга. В атрофированных участках коры отмечаются уменьшение количества нервных клеток, пролиферация фиброзной глии, отложение кальция.
Патогенез синдрома Стержа-Вебера
Согласно мнению большинства исследователей, синдром Стерджа - Вебера является врожденной мальформацией мезодермальных и эктодермальных элементов головной части эмбриона, возникшей под воздействием различных причин как экзогенных, так и генетически обусловленных. Довольно часто отмечается комбинация синдрома Стерджа - Вебера с другими формами факоматозов, а также абортивные, не полностью выраженные случаи болезни.
Клиника синдрома Стержа-Вебера
Сосудистые пятна на лице носят врожденный характер. Они обычно располагаются на одной стороне лица, в надглазничной области. Распространенность пятен очень варьирует. Сосудистые пятна плоские, красного или вишнево-красного цвета, бледнеют ори надавливании.
Раннее развитие детей, рожденных с подобным пятном, в большинстве случаев нормальное. Однако уже в конце первого или в начале второго года жизни появляются эпилептические припадки. Они могут развиваться постепенно, начинаясь с одиночных или групповых судорог, но могут возникать и в виде эпилептического статуса, как правило, джексоновского типа. Судорожные подергивания развиваются в конечностях, контралатеральных по отношению к расположению пятен на лице. После каждого приступа могут наблюдаться преходящие гемипарезы, выраженность которых нарастает с течением времени. Нередко отмечается отставание в росте конечностей на пораженной стороне. Довольно часто уже в раннем детском возрасте развивается гемианопсия, нередко имеют место глаукома или гидроцефалия. С течением времени нарастает умственная отсталость.
Рентгенологически можно обнаружить обызвествление в затылочных и теменных отделах мозга. При ПЭГ у большинства больных отмечаются атрофия вещества мозга, расширение субарахноидальных щелей на конвекситальной поверхности и расширение полостей желудочков мозга.
Диагноз синдрома Стерджа - Вебера не представляет трудностей. Наличие корковых обызвествлений можно выявить при рентгенографии. Характерные тени располагаются в виде двойных контуров, повторяющих извилины пораженных долей мозга. Компьютерная томография выявляет более обширные области обызвествления, чем это видно при обычной рентгенографии.
Лечение носит симптоматический характер. Проводится регулярное и настойчивое лечение противосудорожными средствами. В некоторых случаях для ликвидации судорожных припадков прибегают к хирургическому лечению, удалению пораженных долей мозга.
Заболевание относится к группе факоматозов. Факоматозы представляют собой наследственные заболевания, характеризующиеся изменениями кожных покровов, наиболее частыми из которых являются пятна (phacos - пятно), неврологическими нарушениями и патологией внутренних органов. Такая комбинация связана с нарушением развития одновременно двух зародышевых листков - эктодермы и мезодермы из этих зародышевых зачатков формируются нервная система кожа и ее производные, внутренние органы и сосуды. К этой группе заболеваний относятся энцефалотригеминальный ангиоматоз Штурге-Вебера, туберозный склероз Бурневилля, синдром Луи-Бар (атаксия-телеангиэктазия), нейрофиброматоз Реклингхаузена. Многие формы патологии передаются из поколения в поколение. Общей клинической закономерностью для заболеваний этой группы является неуклонное прогрессирование симптомов, нередко приводящее к тяжелым необратимым изменениям многих функций организма. Классическое проявление болезни характеризуется триадой симптомов: сосудистыми пятнами на коже лица (ангиомы), судорожными приступами (чаще парциальные), повышением внутриглазного давления (глаукома).
Пигментный невус у пациента с болезнью Штурге - Вебера
Патология часто носит семейный характер с аутосомно-доминантным наследованием, но описаны и аутосомно-рецессивные формы. Ангиомы на коже обычно обнаруживаются уже при рождении, имеют вид «пылающего пятна». В 80% случаев они располагаются на лице (с одной или двух сторон) в области иннервации ветвей тройничного нерва. Судорожные припадки появляются в первые годы жизни, обычно носят очаговый характер. У многих больных они заканчиваются генерализованным судорожным припадком. Возможны бессудорожные приступы в виде мгновенных отключений сознания, вздрагиваний, застываний. У некоторых больных возникают сильные приступы головной боли с рвотой (мигренеподобные приступы). Повышение внутриглазного давления (глаукома) наблюдается с рождения или появляется позднее. Частота появления глаукомы составляет около 50%. Прогрессирование глаукомы приводит к снижению зрения вплоть до полной слепоты. Из других проявлений болезни часто встречается слабоумие, которое обусловлено повторяющимися приступами. Оно сочетается с выраженными изменениями в эмоционально-волевой сфере: злопамятностью, эгоцентризмом, аффективностью, мстительностью. Ухудшаются память, внимание, способность усваивать новые сведения. Указанные факторы значительно осложняют процесс обучения и воспитания. Эти нарушения психики отмечаются и в период между приступами. Выраженность расстройств интеллекта, особенно памяти, нарастает по мере прогрессирования заболевания и учащения судорог. Диагноз энцефалотригеминального ангиоматоза подтверждается при измерении внутриглазного давления, исследовании глазного дна, рентгенографией черепа и записи биотоков мозга.
Компьютерная томография пациента с болезнью Штурге - Вебера
Магнитная резонансная томография пациента с болезнью Штурге - Вебера
При патоморфологическом исследовании больных, страдавших энцефалотригеминальным ангиоматозом, выявляется разрастание сосудов кожи, мягкой мозговой оболочки, сосудистых сплетений глазного яблока. Реже ангиомы локализуются в затылочной области больших полушарий, мозжечке, спинном мозге и внутренних органах. Описано отложение солей кальция в сосудах мозга (петрификаты). Выраженность ангиом может значительно варьироваться. При лечении заболевания используют противосудорожные и психотропные средства, а также препараты, снижающие внутричерепное и внутриглазное давление.
Показанием к нейрохирургической операции является наличие некупируемых приемом противосудорожных препаратов судорог. В таком случае может быть выполнена резекция части очагов или гемисферэктомия.
Синдром Sturge-Weber.
Продолжение.
Не плохая ссылка -
http://jnnp.bmj.com/content/39/5/429.long
http://www.nejm.org/doi/full/10.1056/NEJMicm1700538