Синдром МELAS - заболевание, обусловленное точковыми мутациями в митохондриальной ДНК. Возраст начала заболевания широко варьирует от младенческого до взрослого, однако наиболее часто первые признаки обнаруживаются в возрасте от 5 до 15лет. В большинстве случаев болезнь дебютирует инсультоподобными эпизодами, злокачественными мигренями или задержкой психомоторного развития. Наиболее часто инсульты локализуются в височной, теменной или затылочной долях головного мозга, сопровождаются гемипарезом и гемианопсиеи и имеют тенденцию к быстрому восстановлению.
Причиной возникновения инсультов является митохондриальная ангиопатия, характеризующаяся избыточной пролиферацией митохондрий в стенках артериол и капилляров сосудов мозга. По мере прогрессирования заболевания, на фоне рекуррентного характера инсультов нарастает неврологическая симптоматика - у бального возникают мышечная слабость, судороги, миоклонии, атаксия и нейросенсорная тугоухость. В небольшом проценте случаев помимо неврологической симптоматики возникают эндокринные расстройства в виде сахарного диабета и гипофизарного нанизма.
Диагностика этого заболевания, также как и при СКС и синдроме Пирсона, проводится с использованием биохимического, морфологического и молекулярно-генетического методов. Наиболее частой мутацией в мтДНКявляется однонуклеотид-ная замена А ~> G в 3243-ем положении. Эта мутация приводит к инактивации транскрипционного терминатора, заключенного внутри гена тРНК Leu. Таким образом, последствием этой мутации является изменение транскрипционного соотношения рРНК. и мРНК и снижение эффективности трансляции. Вторая по частоте мутация, приводящая к возникновению синдрома MELAS - замена Т -> С в 3271-ом положении мтДНК.

Генетика: У большинства пациентов (80%) синдром MELAS обусловлен точковой заменой A3243G в гене тРНК лейцина (UUR), 7,5% - точковой заменой Т3271С, еще реже - заменой А3252G в том же гене мтДНК и точковой мутацией в гене COX III. Злокачественная мигрень с инсультоподобными состояниями может быть обусловлена спорадическими делециями мтДНК.
Тип наследования: материнский
Эпидемиология:Точная частота заболевания не известна. В литературе имеются единичные данные о частоте заболевания. На севере Финляндии частота мутации A3243G, составляет 16.3:100 000.
Патогенез: Мутации мтДНК, контролирующих дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования - важнейшего источника энергии для метаболических процессов в клетке. Точковые мутации мтДНК, затрагивающие функцию тРНК, поражают синтез практически всех белков, кодируемых мтДНК, приводя к их сочетанной недостаточности. При мутациях мтДНК чаще всего наблюдается сочетанной ферментной недостаточностью (например, комплексов I+III+IV), причем эта недостаточность вариабельна, но всегда частичная, иногда почти граничит с нормой. Это объясняется двойным генетическим контролем - ядерным и митохондриальным - окислительного фосфорилирования
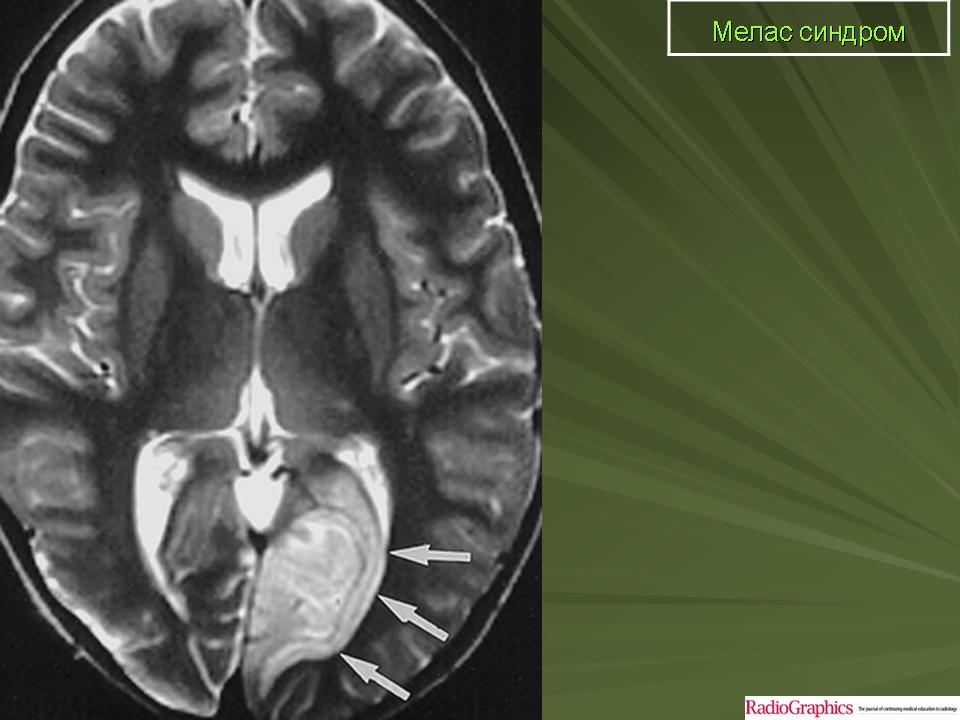
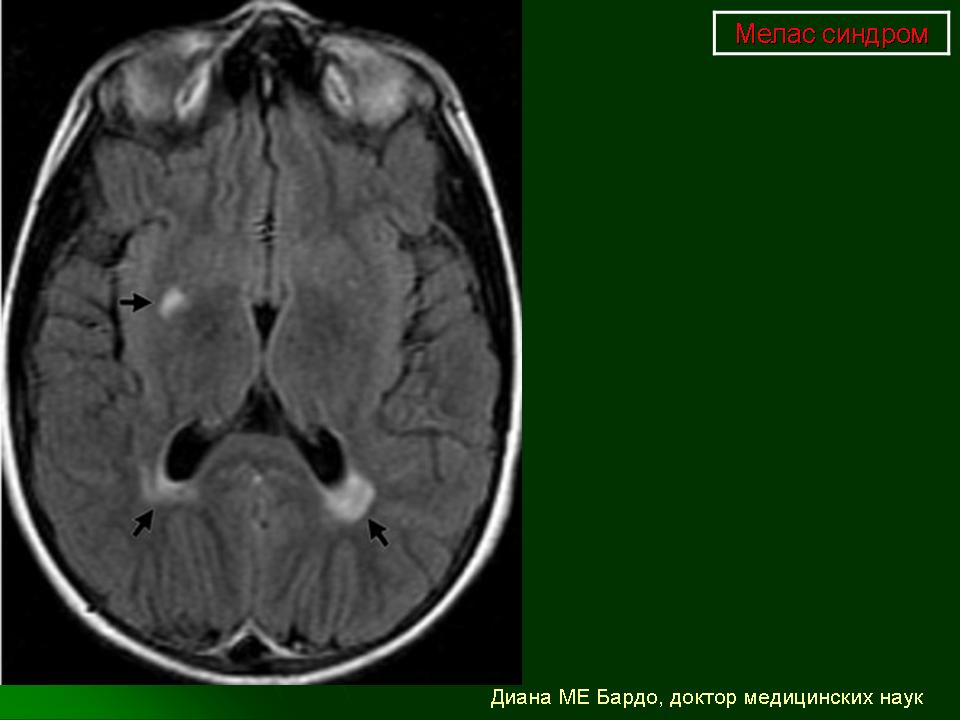
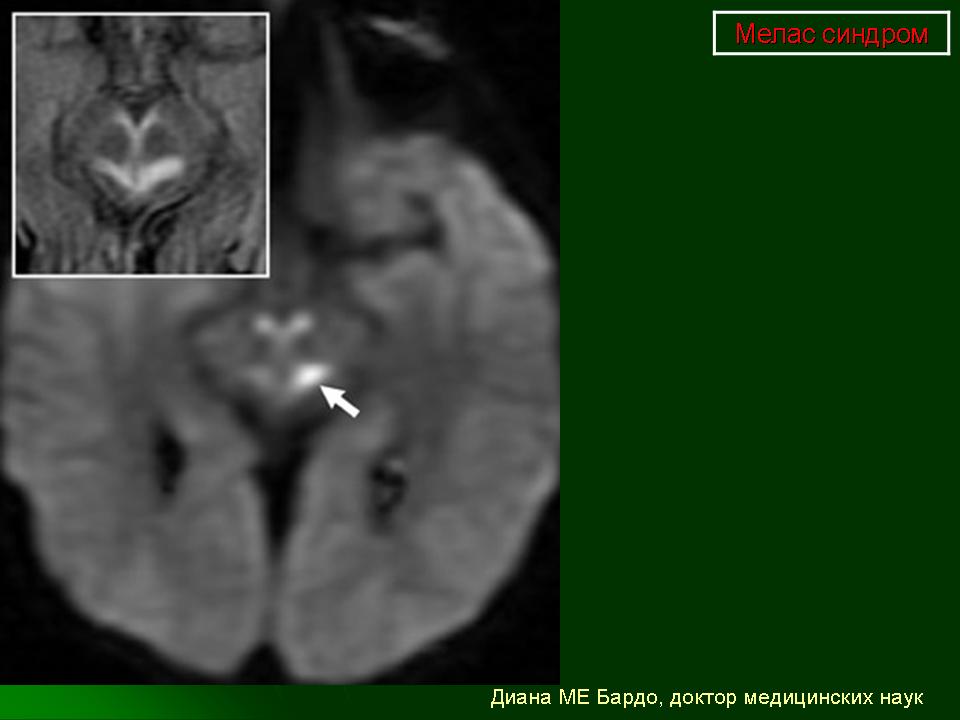
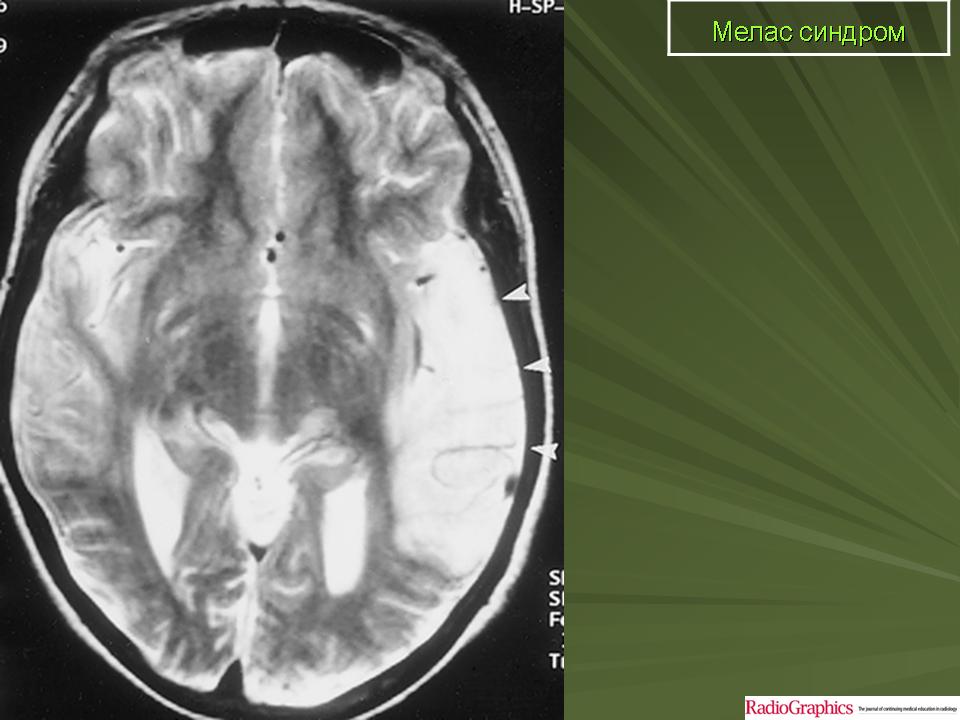
Клинические проявления: Синдром MELAS как правило дебютирует в возрасте от 5 до 35 лет. Заболевание манифестирует либо инсульто-подобными состояниями (кортикальный или субкортикальный инфаркт), либо злокачественной мигренью. Инсульто-подобные состояния чаще всего развиваются в возрасте 5-15 лет. Типичная локализация очагов, выявляемая при проведении КТ/МРТ головного мозга, - височная, теменная или затылочная область. В большинстве случаев они сопровождаются гемипарезами и гемианопсией и имеют тенденцию к относительно быстрому восстановлению при выраженном рекуррентном характере. Основными неврологическими симптомами являются: различные эпилептические приступы ( фокальные, вторично генерализованные, диалептические и другие), мозжечковые расстройства, миоклонус-эпилепсия, корковая агнозия, мигренеподобными головными болями и подкорковыми нарушениями (мышечная дистония, различные виды гиперкинезов). Нейропатологические изменения головного мозга включают гибель нейронов, демиелинизацию, пролиферацию астроцитов. Инсульто-подобные состояния никогда не являются следствием тромбоэмболии, а возникают вследствие митохондриальной ангиопатии и не соответствуют локализации магистральных сосудов мозга. При аутопсии в стенках мозговых артериол и капилляров, в эндотелиальных и гладкомышечных клетках, выявлена гиперпролиферация митохондрий что и обуславливает ангиопатию. Кальцификаты в области базальных ганглиев, нередко встречается при MELAS. Периферическая невропатия наблюдается редко. Мышечная слабость, нейросенсорная тугоухость являются типичными симптомами заболевания. Эндокринопатии могут быть представлены недостаточностью гормона роста, сахарным диабетом. К числу редких симптомов относятся нарушения сердечной проводимости, кардиомиопатия, почечно-канальцевая тубулопатия. Клиническая картина заболевания у пораженных родственников по материнской линии может варьировать от практически бессимптомной до мягко выраженной, включая только тугоухость, отставание роста, мигренеподобные головные боли.
Диагностика: У всех пациентов с синдромом MELAS выявляют повышение лактата и пирувата в крови и ЦСЖ, как в покое, так и после физической нагрузки. Наиболее частой мутацией, приводящей к этому синдрому является замена A3243G. Эта мутация встречается у 80% больных с данным синдромом. Мутация G13513A является второй по частоте среди пациентов европейского происхождения. Диагностика проводится в лаборатории наследственных болезней обмена веществ МГНЦ РАМН (http://www.labnbo.narod.ru). Морфологическое исследование мышечного биоптата проводиться в Московском научно-исследовательском институте педиатрии и детской хирургии (http://www.pedklin.ru )
Лечение: В настоящее время, специфического лечения для лечения MELAS не разработано, проводится симптоматическая терапия. Всем больным проводится нейротрофическая, метаболическая терапия, как и при всех митохондриальных заболеваниях. Одним из вариантов лечения рассматривается внутривенное введение L-аргинина.
Прогноз
Прогноз по заболеванию неблагоприятный.
С.К. Евтушенко, Ю.М. Перепечаенко, Е.М. Соловьева, А.В. Душацкая, Донецкий государственный медицинский университет, Украина
Международный неврологический журнал 4(8) 2006 / Практикующему неврологу /To practicing neurologist/
Синдром MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke) — митохондриальная энцефаломиопатия, лактат-ацидоз и инсульт — выделен сравнительно недавно (в 1984 г.). Заболевание связано с точковой мутацией митохондриальной ДНК, которая в 90% локализуется в гене, кодирующем синтез транспортной РНК лейцина, что препятствует его включению в белки дыхательной цепи. Как и при всех митохондриальных заболеваниях, диагностика синдрома MELAS представляет трудности, обусловленные значительным разнообразием клинической картины.
Основные клинические проявления синдрома MELAS: непереносимость физических нагрузок; рецидивирующие инсультоподобные состояния. Термин «инсультоподобные», вероятно, обусловлен тем, что в большинстве описываемых случаев ведущим клиническим проявлением является головная боль с рвотой, судорогами, часто с нарушением сознания, длительностью от нескольких часов до нескольких дней.
Во время этих атак у ряда больных нередко развиваются неврологические нарушения в виде гемианопсии, гемипареза, редко в виде афазии. При проведении КТ у 60% таких пациентов выявляются очаги пониженной плотности, полиморфные судорожные приступы; при биохимическом исследовании выявляется лактат-ацидоз; начало заболевания — в 5-6-летнем возрасте; течение болезни носит прогрессирующий характер.
Приводим собственное наблюдение MELAS синдрома.
Больной А., 6 лет, поступил в клинику с жалобами на остро развившуюся слабость в правых конечностях, которая удерживалась в течение нескольких часов, головные боли. Родился от первой беременности. Беременность и роды протекали без осложнений. Раннее психомоторное развитие ребенка соответствовало возрасту. С 1 года до 2 лет отмечались аффективно-респираторные пароксизмы. С 3 лет частые ацетонемические состояния, развивающиеся на высоте головной боли.
Во время осмотра в клинике: двигательная расторможенность, неусидчивость, быстрая утомляемость при нагрузках (как умственных, так и физических). В неврологическом статусе: черепная иннервация без особенностей, мышечная гипотония, асимметрия сухожильных рефлексов (более живые справа). Парезов нет. Атаксии нет.
При первичном проведении МРТ головного мозга в левой затылочной доле обнаружена обширная зона гиперинтенсивного сигнала в режиме Т2 и гипоинтенсивность в режиме Т1 с четкими контурами, срединные структуры не смещены. Данная радиологическая картина была расценена как явление ишемического инсульта.
В течение последующего года у ребенка трижды отмечались парциальные эпилептические приступы, повторялись эпизоды приступообразной головной боли с рвотой. Получал противосудорожную терапию. Спустя год после первого лечения в клинике мальчик поступил повторно в связи с развившимся приступом генерализованных тонико-клонических судорог, который в течение часа повторился дважды.
Наросла вялость, слабость, появилась приступообразная головная боль с однократной рвотой. Повторно проведена МРТ головного мозга — в режимах Т1 иТ2 визуализировались участки измененного сигнала в теменно-затылочных отделах с обеих сторон (причем очаг слева имел меньшие размеры по сравнению с таковым при предыдущем МРТ-исследовании). Таким образом, за прошедшие после первого инсульта несколько месяцев мальчик перенес по крайней мере еще три острых нарушения мозгового кровообращения.
Метаболические нарушения заключались в значительном увеличении уровня лактата в крови до 4,2 ммоль/л (норма до 1,7 ммоль/л) и пирувата.
Комплексный анализ результатов обследования и клиники позволил установить конкретную нозологическую форму митохондриальной энцефаломиопатии — синдром MELAS, который в клинике был установлен впервые.
В последующем ребенок получал противосудорожную терапию (топамакс 5 мг/кг/сут.), лечение, направленное на стимуляцию тканевого дыхания с использованием коэнзима Q-10, янтарной кислоты, цитомака, а также актовегин и контрикал.
Приведенный случай рецидивирующих метаболических инфарктов мозга у ребенка, обусловленных синдромом MELAS, не является единственным случаем в клинике, и банк данных о подобных больных накапливается.
Синдром МELAS
Ш.Ш. Шамансуров, Ш.Х. Саидазизова, Ташкентский институт усовершенствования врачей, Узбекистан
В последние годы анализ причин нарушений нервно-психического развития показывает, что определенная доля принадлежит группе заболеваний, вызываемых дефектами структуры и функции митохондрий, т.е. митохондриальным заболеваниям.
Функциональная и структурная недостаточность митохондрий вызывает энергетический дефицит клеток. Митохондриальные заболевания характеризуются поражением ЦНС, низкой переносимостью физических нагрузок, мышечной слабостью.
Диагностика митохондриальных заболеваний представляет определенные трудности в силу необходимости использования сложных аналитических методов, но при тщательно собранном анамнезе, учете генеалогических особенностей, фенотипических признаков можно заподозрить заболевания митохондриального генеза.
Митохондриальные заболевания могут возникать в результате:
1) точковой мутации митохондриальной ДНК (наследование по материнской линии);
2) делеции или дупликации митохондриальной ДНК (не наследуется);
3) множественной митохондриальной делеции;
4) деплеции — отсутствия или уменьшения числа копий митохондриальной ДНК в тканях.
Таким образом, многообразие способов наследственной передачи заболеваний актуализирует необходимость тщательного сбора анамнеза, изучения генеалогических особенностей и подробного клинического и клинико-нейрофизиологического обследования аналогичных больных.
Начальные симптомы митохондриального заболевания могут появиться с первых дней жизни с последующим прогрессированием течения. Трудность ранней диагностики заключается в том, что специфическая симптоматика проявляется не сразу после манифестации начальных признаков, а спустя некоторое время, и заболевание характеризуется исключительным разнообразием симптомов и сочетанным поражением различных органов.
Клинически митохондриальные заболевания проявляются миопатическим синдромом, поражением нервной системы, поражением сердца, печени, почек, эндокринными нарушениями, нарушениями слуха, зрения.
В нашей клинике на протяжении последних лет стала прослеживаться тенденция к постановке диагнозов, указывающих на наличие митохондриального поражения.
Один из таких случаев митохондриальной патологии — синдром MELAS. В литературе этот синдром трактуется как митохондриальная энцефалопатия с инсультоподобными эпизодами.
Митохондриальная энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды (синдром MELAS) впервые выделены в самостоятельную нозологическую единицу в 1984 году.
Частота не установлена, но к 1998 году опубликовано более 120 наблюдений (П.А. Темин и др., 1995; Е.А. Николаева и др., 1997).
В основе данной патологии лежит точковая мутация митохондриальной ДНК, вызывающая нарушение продукции рибосомальной РНК и дефицит энергетической продукции митохондриальной дыхательной цепи.
У больных с синдромом MELAS содержание аномальной митохондриальной ДНК в различных тканях составляет 93–96 %. У членов семьи пробандов в тканях также определяется мутантная ДНК, но ее содержание существенно ниже: 62–89 % при стертой форме болезни, от 28 до 89 % при отсутствии клинических признаков синдрома (П.А. Темин, Л.З. Казанцева, 2001).
Заболевание наследуется по материнской линии с высоким риском. Но по данным литературы известно, что лишь у 25–44 % больных наблюдается отягощенный семейный анамнез, в остальных случаях заболевание в родословной регистрировалось впервые.
В условиях нашего стационара с 2001 года наблюдается больная С.Н., 14 лет, впервые обратившаяся с жалобами на судороги, общую слабость, утомляемость, депрессивное настроение, непереносимость физических нагрузок. За 5 лет наблюдений отмечается периодическое прогрессирование симптомов с инсультоподобными эпизодами.
В родословной по линии матери пробанда отмечаются случаи патологии, которые можно охарактеризовать как энцефаломиопатию, эпилепсию. Мать пробанда страдает синдромом сахарного диабета с тугоухостью и отмечает периодическую мышечную утомляемость.
Анамнез жизни. Девочка от второй беременности, 1-х родов. 1-я беременность закончилась невынашиванием. Данная беременность протекала на фоне соматической ослабленности матери. Акушерский анамнез отягощен: имела место слабость родовой деятельности и оказывались мероприятия по стимуляции родового акта. Масса тела при рождении — 3200 г. Закричала сразу. К груди приложена на 2-е сутки.
Анамнез болезни. Под наблюдением неврологов ребенок находился с 3-месячного возраста по поводу перинатальной энцефалопатии. Относится к группе часто болеющих детей. С 3–4-летнего возраста у ребенка констатируется хронический тонзиллит. С 6–7-летнего возраста замечено отставание физического развития, по поводу чего наблюдались у эндокринолога. С 12-летнего возраста девочка страдает судорожным синдромом, впервые возникшим на фоне вирусной инфекции. Судороги носят парциальный характер и сопровождаются вегетативными нарушениями в виде гипергидроза, тошноты, чувства страха. Судороги резистентны к терапии.
Объективно: состояние при поступлении тяжелое. Дефицит роста — 10 см, массы тела — 15 кг. Больная вяла, гиподинамична, контактна, но отмечается навязчивость мышления, обстоятельность, педантичность.
В соматическом статусе: кожные покровы бледные, подкожно-жировой слой развит слабо. В легких везикулярное дыхание. Границы сердца не расширены. Тоны приглушенные, ритмичные, умеренная тахикардия (ЧСС — 90–100 уд./мин), короткий систолический шум в точке Боткина. Живот мягкий, безболезненный. Печень и селезенка не увеличены. Симптом Пастернацкого отрицательный.
В неврологическом статусе: лицо гипомимично, углы губ опущены, выражение лица скорбное, плечи опущены. Дизартрия, смазанность речи с легким гнусавым оттенком. Полуптоз слева. Снижение конвергенции слева. Горизонтальный нистагм при крайних отведениях глазных яблок с обеих сторон. Глоточный рефлекс снижен. На фоне диффузной мышечной слабости выявляется правосторонний гемипарез центрального типа с гиперрефлексией, клонусом стопы, патологическим рефлексом Бабинского. Координаторные пробы: интенция, мимопопадание при пальценосовой пробе с обеих сторон. Выражена атаксия. В позе Ромберга неустойчива, отмечается ретро- и латеропульсия.
Выявлен миопатический синдром, проявляющийся в слабости и атрофии мышц, снижении мышечного тонуса, мышечных болях (крампи). Больная не переносит физических нагрузок.
С использованием таблицы, в которой приведены начальные признаки синдрома MELAS, был проведен анализ клинических симптомов, обнаруженных нами у пробанда.
В табл. 1 и 2 показано, какие из начальных и других симптомов синдрома MELAS выявлены у пробанда в ходе обследования.
Были проведены консультации узких специалистов.
Отоларинголог: правосторонняя нейросенсорная тугоухость 1–2-й степени.
Офтальмолог: VIS OD — 0,9; VIS OS — 0,8. Признаки венозного застоя ДЗН. Ангиопатия сетчатки.
Эндокринолог: задержка физического и полового созревания. Гиперпролактинемия.
Данные лабораторных и функциональных исследований
ЭЭГ: очаг судорожной активности, исходящий из стволовых структур, на фоне сниженной биоэлектрической активности головного мозга.
Допплерография экстра- и интрацеребральных сосудов: признаки внутричерепной гипертензии с артериоспазмом, больше справа. Дефицит скорости кровотока по основной артерии.
МРТ головного мозга: гиподенсивный очаг в проекции теменной области справа — ОНМК по ишемическому типу. Энцефалопатия. Субатрофия головного мозга с признаками эктазии желудочков.
Электрокардиография: признаки обменных нарушений, неполная блокада правой ножки пучка Гиса.
При общем анализе крови выявлена гипохромная анемия 1 степени.
Биохимический анализ крови: АЛТ —2,36 ммоль/л; общий билирубин — 76,3 ммоль/л; СА крови — 2,24 ммоль/л.
Анализ крови на выявление лактат-ацидоза — положительный (абсолютный признак).
Анализ мочи: органическая ацидурия с экскрецией молочной и пировиноградной кислот.
Биопсия мышечной ткани (окраска трихромом по Гомори): «рваные» красные волокна.
Комплексный анализ результатов обследования пробанда позволил установить у ребенка одну из нозологических форм митохондриальной энцефаломиопатии — синдром MELAS.
Доказательства:
— наличие у матери и родственников по материнской линии клинических признаков патологии типа митохондриальной энцефаломиопатии;
— манифестация болезни после 6-летнего возраста;
— прогрессирующий характер заболевания;
— особенности клинической симптоматики.
Ребенку кроме посиндромного, симптоматического лечения была назначена терапия, направленная на стимуляцию процессов тканевого дыхания в виде комплекса препаратов коэнзима Q10, лецитина. Проведено внутривенное капельное введение иммуноглобулина человеческого № 3. Назначена плановая терапия антиконвульсантами. Через 1 месяц после проведенного лечения отмечалась существенная положительная динамика клинического состояния. Прекратились судороги (выраженный позитивный сдвиг на ЭЭГ в виде дисфункции подкорковых структур), пациентка стала реже и легче переносить простудные заболевания, прекратились головные боли, приступы сонливости, исчезли крампи, уменьшилась выраженность птоза. Самостоятельно ходит. Улучшилось настроение и контакт с окружающими.
Из представленного можно сделать следующие выводы: констатация митохондриального поражения требует более тщательного подхода к лечению с включением в комплекс терапии препаратов метаболического действия, улучшающих процессы тканевого дыхания, окислительного фосфорилирования в клетках. Только регулярное проведение системной терапии помогает поддерживать состояние больных и предотвращать повторение эпизодов инсульта.
http://www.neurologyindia.com/article.asp?issn=0028-3886;year=2010;volume=58;issue=5;spage=791;epage=793;aulast=Aaron