Синдром Пьера Робена у детей.
Кириллова Л.Г., Ткачук Л.И., Шевченко А.А., Силаева Л.Ю., Лисица В.В., Мироняк Л.А., ГУ «Институт педиатрии, акушерства и гинекологии АМН Украины», Научно-практический центр лучевой диагностики АМН Украины, г. Киев
На сегодняшний день в Украине к наиболее распространенным заболеваниям у детей относятся неврологические расстройства, частота которых в последние годы, к сожалению, значительно возросла. Данные нашей клиники свидетельствуют и об изменении их структуры с увеличением нейрогенетической, нейрометаболической патологии и имитируемых расстройств, а также увеличением частоты врожденных пороков центральной нервной системы (ЦНС).
Прошло более семи лет с тех пор, как была проведена международная научно-практическая конференция в г. Святогорске (Донецкая область) по неординарным (раритетным) заболеваниям нервной системы у детей и взрослых. В материалах этой конференции были освещены малоизученные на то время, редкие, труднодиагностируемые заболевания. По выражению проф. С.К. Евтушенко, «долго остаются неизвестными редкие болезни, пока их не увидит опытный клиницист» [1]. С течением времени мы пришли к выводу о необходимости проведения повторной конференции, но уже с участием врачей различных специальностей, целью которой будет обсуждение редких и мало выявляемых заболеваний у детей, что позволило бы повысить уровень профессиональной подготовки практикующих врачей и способствовало бы улучшению качества оказания помощи больным.
В последние 5 лет в клинике детской психоневрологии нашего института были диагностированы такие необычные и редкие патологические неврологические нозологии, как атрофия зрительных нервов Лебера, синдром Пьера Робена, синдром Кернса - Сейра, синдром Толоза - Ханта, синдром Клиппеля - Треноне, синдром Прадера - Вилли, синдром Мюнхгаузена, липогранулематоз Фарбера, нарушение метаболизма креатина как причина развития резистентной формы детской эпилепсии и т.д., что нашло отражение в ряде соответствующих публикаций [2-6].
В настоящей работе на страницах уважаемого международного неврологического издания мы хотим представить случай синдрома Пьера Робена у ребенка с микрогнатией и глоссоптозом в сочетании с поражением центральной нервной системы в виде структурных изменений мозга и клинических проявлений (мышечная гипотония, задержка психомоторного развития, тяжелые эпилептические припадки, отсутствие слуховых и голосовых функций).
Согласно данным литературы, дефекты центральной нервной системы у больных с синдромом Пьера Робена составляют около 50 % случаев [19]. Однако в доступных нам источниках мы не встречали описаний синдрома Пьера Робена в сочетании с грубыми деструктивными изменениями в головном мозге и подтверждением дисфункции слуховых труб (на МРТ головного мозга), которая может быть обусловлена секреторным отитом.
В настоящее время только в единичных работах приведены эпидемиологические данные по этому синдрому [13, 19]. Согласно результатам ретроспективного исследования, распространенность этой патологии в Дании составляет 1 случай на 14 000 живорожденных детей; по данным американского исследования - 1 случай на 2-30 тысяч. Соотношение мужчин и женщин - 1 : 1 [13]. В Украине точных данных о распространенности синдрома Пьера Робена на сегодняшний день нет. За последние годы в клиниках нашего института наблюдалось 7 пациентов. Можно предположить, что данная патология встречается в популяции довольно часто, но выявляемые признаки не рассматриваются в совокупности как проявления единого заболевания.
Сведения о нарушении развития нижней челюсти и неба, сочетающемся с нарушением дыхания, появились в медицинской литературе еще в 20-е годы ХІХ века. В 1923 году французским стоматологом Пьером Робеном была установлена связь между гипоплазией нижней челюсти (микрогнатией), расщелиной мягкого неба и обструкцией дыхательных путей, которая состоит в последовательном появлении симптомов друг за другом. В дальнейшем патология была названа именем этого ученого, и в зарубежной литературе используется термин «последовательность Пьера Робена» [20].
Нарушение эмбрионального развития нижней челюсти происходит как при наличии механической компрессии внутри матки, так и при воздействии инфекции на ранних этапах беременности или нейрогенетических нарушениях. Следует отметить, что последовательность Пьера Робена может быть изолированным синдромом или проявлением генетической патологии. Как часть генетической патологии последовательность Пьера Робена описана почти при 300 синдромах. Наследование может происходить и по аутосомно-доминантному, и по аутосомно-рецессивному типу. Вероятность рождения больного в семье, уже имеющей ребенка с синдромом Пьера Робена, составляет 1-5 % [9, 10].
В клинической картине заболевания прежде всего обращает на себя внимание микрогнатия, приводящая к патологической ретрогнатии (смещение нижней челюсти назад) и уменьшению объема ротовой полости. Как следствие, язык смещается в сторону задней стенки глотки, возникает глоссоптоз, что наблюдается в 75-80 % случаев. Сочетание микрогнатии и глоссоптоза вызывает серьезные затруднения дыхания и питания у новорожденного, вследствие чего часто развивается инспираторная обструкция дыхательных путей.
В зависимости от состояния дыхания выделяют три степени тяжести синдрома Пьера Робена:
1) легкая степень: дыхание не затруднено, незначительные трудности с кормлением, устраняемые консервативным лечением в aмбулаторных условиях;
2) средняя степень: умеренное затруднение дыхания, умеренные трудности с кормлением, требующие лечения в стационаре;
3) тяжелая степень: значительное затруднение дыхания, значительные трудности с кормлением в течение длительного времени, приводящие к необходимости использования дыхательного пособия в виде интраназального зонда в нижние дыхательные пути или трахеостомии [7, 12, 14].
Хотим обратить внимание на то, что согласно описаниям разных авторов при синдроме Пьера Робена патология ротовой полости сочетается с пороками развития других органов и систем. Наиболее частыми являются аномалии слухового аппарата (75 % случаев) с потерей слуха у 60 % больных [19, 21]. Кроме того, описаны: врожденная катаракта, миопия, пороки сердца, мочеполовой системы, аномалии развития грудины и позвоночника, а также полидактилия и врожденное отсутствие конечностей [8, 19, 21]. Дефекты центральной нервной системы составляют около 50 % случаев у пациентов с синдромом Пьера Робена и включают в себя: задержку развития психики и речи, двигательные нарушения, микроцефалию или гидроцефалию [21, 22]. Частыми бывают эпилептические припадки. Умственная отсталость отмечается примерно у 20 % больных [11, 15, 16].
Согласно данным зарубежных авторов, при проведении высококачественного ультразвукового исследования диагноз синдрома Пьера Робена должен быть определен пренатально [7]. После рождения патология диагностируется в первый день жизни на основании данных пренатальной УЗ-диагностики и клинической картины. Характерен внешний вид новорожденного - так называемое птичье лицо, что связано с недоразвитием нижней челюсти (от слабо выраженного недоразвития до полного ее отсутствия). В разные сроки, в зависимости от степени тяжести порока, появляется резкое нарушение дыхания, связанное с западением языка, возможны частые апноэ. В таких случаях ребенок беспокоен, выражен цианоз кожных покровов. Из-за нарушения акта глотания во время кормления ребенка может наступить удушье.
Анализируя данные литературы, мы хотим акцентировать внимание на осложнениях при синдроме Пьера Робена:
1) обструкция верхних дыхательных путей, которая вызывает стридорозное дыхание, ларингомаляцию, а в тяжелых случаях - асфиксию во время сна;
2) отставание в физическом развитии;
3) отставание в психомоторном развитии;
4) нарушение речи;
5) хронические инфекционные заболевания уха, ведущие к потере слуха;
6) ортодонтические проблемы;
7) апноэ, смерть во время сна.
Лечение детей с синдромом Пьера Робена подразумевает прежде всего устранение обструкции верхних дыхательных путей и расщелины мягкого неба, коррекцию кормления. В течение всего периода раннего детства пациенты должны регулярно наблюдаться отоларингологом, офтальмологом, логопедом и ортодонтом. Подходы к лечению включают консервативные и хирургические методы.
В 80 % случаев легкой и средней степени тяжести эффекта можно достичь применением позиционной терапии - обязательного укладывания детей на живот. При средней и тяжелой степенях необходимо использование интраназального зонда в верхние дыхательные пути.
Хирургическое лечение состоит во временной глоссопексии - подтягивание языка вперед с фиксацией к нижней губе, а в крайне тяжелых случаях и трахеостомии.
Важным представляется, что устранение расщелины мягкого неба должно быть проведено до становления речи, поэтому операции производятся в возрасте 6-19 месяцев в зависимости от степени дыхательной обструкции [14, 17, 18].
Ниже представляем историю болезни пациента с синдромом Пьера Робена, находившегося на лечении в отделении детской психоневрологии ГУ «ИПАГ АМН Украины». Ребенок Р., мальчик, в возрасте 1,5 месяца поступил в отделение детской психоневрологии с жалобами на задержку в развитии - взгляд не фиксирует, не реагирует на звуки, не кричит, самостоятельно не сосет (питание получает через зонд, вводимый матерью через ротовую полость при каждом кормлении); периодически отмечается западение языка (глоссоптоз); частые вздрагивания.
Ребенок родился от первой беременности, протекавшей с ОРВИ на 2-м месяце, с субфебрильной температурой в течение 3 суток (без медикаментозной терапии), первых срочных родов на 40-й неделе беременности. В родах - затруднение выведения плечиков, ручное пособие по Цовьянову. Родился с массой тела 3400 г, длина 52 см, окружность головы 34 см, в асфиксии тяжелой степени, с оценкой по шкале Апгар 1-3 балла. Сразу после рождения переведен на искусственную вентиляцию легких, продолжавшуюся в течение 4 суток. Состояние ребенка было очень тяжелым: синдром угнетения ЦНС, мозговая кома ІІ-ІІІ степени; наблюдались частые судороги тонико-клонического характера, общая мышечная гипотония, пастозность лица и конечностей, ненапряженная кефалогематома левой теменной кости.
Считаем необходимым обратить внимание на то, что при пренатальном УЗ-исследовании плода (3-кратно по месту жительства) костная патология и нарушение развития структур мозга не были выявлены.
Ребенку дважды в динамике проводилась нейросонография: на 3-и и 14-е сутки жизни. Приводим результаты исследования.
На 3-и сутки: размеры передних рогов боковых желудочков справа - 3,5 мм, слева - 3,2 мм, сосудистые сплетения: справа - 6,7 мм, слева - 7,2 мм, повышенной эхогенности, структурно неоднородные, с неровными контурами; полость прозрачной перегородки - 6 мм; 3-й желудочек - 4,2 мм; межполушарная борозда - 3,2 мм; субарахноидальное пространство - 3 мм. Отсутствует четкая дифференциация структур мозга. Выражено повышение эхогенности паренхимы мозга диффузно (угроза развития кортикальной лейкомаляции).
На 14-е сутки: увеличились размеры передних рогов боковых желудочков мозга до 5,0-5,1 мм, 3-го желудочка - до 6,2 мм; межполушарной борозды - до 5,0 мм. Сохраняется нечеткость контуров основных структур мозга, выраженное диффузное повышение эхогенности паренхимы мозга, ее неоднородность с множественными анэхогенными участками до 5 мм (субкортикальная лейкомаляция?).
На 23-и сутки жизни проведена компьютерная томография головного мозга: желудочки и цистерны головного мозга слабо дифференцируются, за исключением IV, который не расширен и не деформирован. Деструктивных изменений костной ткани не выявлено. Отсутствует дифференциация долей головного мозга справа и слева от уровня средней черепной ямки до верхних отделов желудочков, за исключением левой теменной доли и мозжечка. Основная, фронтальная и гайморова пазухи, ячеистые клетки пирамид и решетки не сформированы.
На фоне лечения, проводимого по месту жительства, состояние ребенка несколько улучшилось (до состояния средней тяжести): появилось самостоятельное дыхание и незначительная двигательная активность. Ребенок самостоятельно не сосал и не глотал, питание получал через назогастральный зонд. Сохранялись мышечная гипотония и угнетение физиологических рефлексов новорожденного. Выписан в месячном возрасте из областной больницы для дальнейшего наблюдения и лечения по месту жительства с диагнозом: «Асфиксия тяжелой степени. Гипоксически-ишемическое поражение ЦНС, тяжелая степень, субкортикальная церебральная лейкомаляция, судорожный синдром, синдром церебрального угнетения. Токсико-гипоксическая кардиопатия, НК І степени. Кераторетинопатия обоих глаз». Рекомендовано питание через назогастральный зонд.
В возрасте 1,5 месяца ребенок поступил в отделение детской психоневрологии ГУ «ИПАГ АМН Украины» с вышеуказанными нарушениями. При первичном осмотре на консилиуме был установлен диагноз: «Синдром Пьера Робена». На рис. 1 представлена фотография данного пациента.
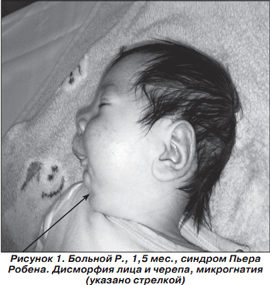
При поступлении состояние ребенка было крайне тяжелым. Почти постоянно отмечались серии инфантильных спазмов, периодически чередующиеся с клоническими и тонико-клоническими судорогами в конечностях. Окружность головы 36 см, окружность грудной клетки - 36 см, большой родничок практически закрыт. Обращала на себя внимание дисморфия лица и черепа, гипоплазия нижней челюсти (микрогнатия), высокое «готическое» небо, язык смещен назад к стенке глотки (глоссоптоз), расщелины мягкого и твердого неба не выявлено. Со стороны черепных нервов: глазные щели D < S, зрачки D = S, умеренно расширены, фотореакции снижены, альтернирующее сходящееся косоглазие, глоточный рефлекс значительно снижен, едва вызывается сильным надавливанием на корень языка. Сосательных и глотательных движений нет. Взгляд не фиксирует, на звук не реагирует, отсутствуют эмоциональные реакции, не кричит, не плачет; при насильственных болевых раздражителях отмечается только незначительная двигательная реакция туловища по типу шевеления. Хватательный и ладонно-ротовой рефлексы отсутствуют. Рефлекс опоры отсутствует, рефлексы автоматической ходьбы и ползания не вызываются. Мышечная гипотония в конечностях, сухожильные и периостальные рефлексы высокие, D = S. Симптом Бабинского с обеих сторон. Температура тела периодически снижается до 34 °С, пульс в пределах 80-
110 уд/мин, дыхание - 32-40 в 1 мин.
Родители ребенка здоровы. Матери 25 лет, отцу - 27 лет. У матери - легкая микрогнатия, рост 158 см.
Проведено обследование: результаты общеклинического обследования в пределах возрастной нормы.
ЭКГ: синусовая аритмия с склонностью к брадикардии. Вертикальное положение электрической оси сердца. В остальном ЭКГ без особенностей.
Эхокардиография: открытое овальное окно. Дефект межжелудочковой перегородки в мембранозной части.
УЗИ органов брюшной полости: без значительных изменений.
Обзорная рентгенография органов грудной клетки: отек межуточной ткани (интерстициальный отек).
Мать и ребенок обследованы на инфекции TORCH-комплекса методом ПЦР - ДНК цитомегаловируса и вируса герпеса I и II типа не обнаружены.
Ребенок консультирован челюстно-лицевым хирургом. Оперативное лечение не показано, рекомендована стимуляция сосательного рефлекса.
Консультация генетиков без существенных дополнительных результатов.
Магнитно-резонансная томография головного мозга выполнена в НДЦ АМН Украины. Приводим подробное описание. На серии MP-томограмм головного мозга белое вещество полушарий большого мозга уменьшено в объеме. В его проекции, а также по ходу мозговых извилин визуализируются множественные энцефаломаляционные полости. На Т1-ВИ в проекции белого вещества, базальных ядер и по ходу проводящих путей мозга отмечаются без четких контуров участки гиперинтенсивного MP-сигнала, которые могут быть обусловлены петрификатами, изменением обменных процессов на фоне нарушения церебрального метаболизма либо последствиями геморрагического пропитывания. Аналогичные по интенсивности зоны невыраженного повышения интенсивности MP-сигнала прослеживаются по ходу зубчатых ядер мозжечка, MP-сигнал от коры мозжечка умеренно повышен на Т2-ВИ. 3-й и боковые желудочки мозга расширены. Расширены подпаутинные пространства над полушариями большого мозга. Мозолистое тело истончено. Образования средней линии не смещены. Отмечается гипоплазия медиальных отделов полушарий мозжечка. 4-й желудочек расположен по средней линии, не изменен. Передняя и средняя мозговая артерии представлены нитевидными сосудами, характерное для их изображения «выпадение» MP-сигнала на Т2-ВИ отсутствует. Зрительные нервы, хиазма, гипофиз, стволовые структуры без особенностей. Отмечается повышение интенсивности MP-сигнала на Т2-ВИ в проекции сосцевидных отростков, барабанных полостей и по ходу слуховых труб, что может быть обусловлено секреторным отитом на фоне дисфункции слуховых труб. Заключение: MP-признаки выраженных деструктивных изменений в полушариях большого мозга, которые могут быть следствием внутриутробной гипоксии либо нейроинфекции.
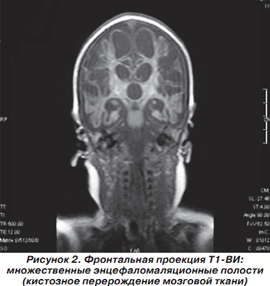
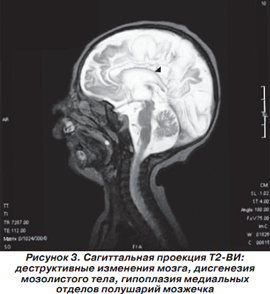
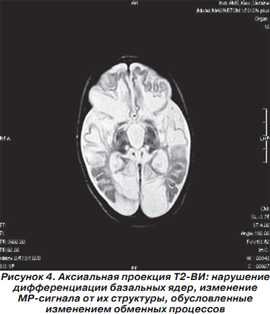
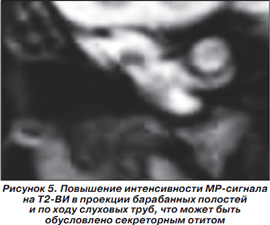
На рис. 2-5 представлены результаты МРТ пациента Р., 1,5 мес., с синдромом Пьера Робена.
На основании всего вышеизложенного установлен заключительный диагноз: «Множественные врожденные пороки развития (ЦНС, костной и сердечно-сосудистой системы) в виде синдрома Пьера Робена с грубыми деструктивными изменениями головного мозга (согласно данным МРТ), эпилептический синдром, выраженная задержка развития двигательных и психоэмоциональных функций, дефект межжелудочковой перегородки, открытое овальное окно».
В результате комплексно проводимой антиконвульсантной (фенобарбитал и клоназепам в сочетании с сернокислой магнезией, сибазон - при статусном течении эпилептических припадков) и нейрометаболической терапии (кортексин, цераксон, пантокальцин, актовегин) частота припадков значительно уменьшилась, появились сосательные и глотательные движения (самостоятельно высасывает до 15 мл грудного молока из соски), глоссоптоза и апноэ в последние дни пребывания в стационаре не наблюдалось, относительно нормализовалась температура тела. Для дальнейшего лечения переведен в областную больницу по месту жительства.
Таким образом, в представленном случае у ребенка имеет место синдром Пьера Робена (микрогнатия, ретрогнатия, глоссоптоз без расщелин твердого и мягкого неба) в сочетании с тяжелым органическим поражением центральной нервной системы в виде грубых структурных изменений мозга (данные МРТ) и клинических проявлений: мышечной гипотонии, задержки психомоторного развития, тяжелых эпилептических припадков, отсутствия слуховых и голосовых функций.
В заключение считаем необходимым обратить внимание на то, что неосведомленность медицинских работников может привести к неправильной тактике ведения таких пациентов. Увидев новорожденного с микрогнатией, всегда следует подумать о синдроме Пьера Робена. В таком случае необходимо тщательно обследовать ротовую полость ребенка с целью выявления расщелины мягкого и твердого неба, а также глоссоптоза. Для предотвращения западения языка и развития апноэ следует с первых дней применять позиционную терапию - выкладывание ребенка на живот в горизонтальном положении.
Продолжение.
Продолжение.
..."Таким образом, в представленном случае у ребенка имеет место синдром Пьера Робена (микрогнатия, ретрогнатия, глоссоптоз без расщелин твердого и мягкого неба)"...
Без расщелины неба - это НЕ синдром Пьера Робена (расщелина неба - обязательный признак этого синдрома). И вся тяжесть состояния этого ребенка обусловлена поражением ЦНС, а недоразвитие нижней челюсти для дыхания в данном случае имеет мало значения - что и было подтверждено (по тексты статьи) применением только медикаментозного лечения.
facedoctor