Клинико-генетические аспекты прогрессирующей оссифицирующей фибродисплазии
В.В. Бадокин, В.А. Мякоткин
рогрессирующая оссифицирующая фибродисплазия (ПОФ) – редкая нозологическая форма, которая характеризуется генетически обусловленной интермиттирующей гетеротопической оссификацией и врожденными дефектами развития, прежде всего скелета. Заболевание характеризуется неуклонно прогрессирующим течением, приводит к значительным нарушениям функционального состояния опорно-двигательного аппарата, глубокой инвалидизации больных и преждевременной их смерти, причем преимущественно в детском и молодом возрасте. Основу ПОФ составляет агрессивная пролиферация фибробластов и формирование воспалительных инфильтратов в сухожилиях, связках, фасциях, подкожных тканях и мышцах, что в конечном итоге приводит к их кальцификации и окостенению. Это заболевание вызывает большой интерес, так как расшифровка его патогенеза может стать ключом к активному воздействию как на регенерацию костной ткани при многих патологических процессах, так и к профилактике гетеротопической оссификации любого генеза.
ПОФ является представителем большой группы заболеваний неизвестной природы, наиболее характерным признаком которых является развитие гетеротопического костеобразования или фибровоспалительных нарушений [1]. Гетеротопический остеогенез встречается при многих нозологических формах. Выделяют локальные травматические, параоссальные и параартикулярные окостенения. Вновь возникшая кость по своим морфологическим признакам ничем существенным не отличается от костной ткани скелета. Костеобразование в мягких тканях может быть причиной нарушения функции опорно-двигательного аппарата. Оно приводит к развитию контрактур, внесуставных анкилозов, снижению силы мышц, нейродистрофическим нарушениям. Размеры этого процесса варьируют от небольших участков оссификации до генерализованного окостенения мышц и сухожилий. Такое костеобразование является частным проявлением метапластического остеогенеза, при котором не участвуют клетки периоста и эндоста.
Прогрессивное мышечное окостенение является самостоятельной нозологической формой, характеризующейся трансформацией соединительной ткани мышечных прослоек, сухожилий и связочного аппарата в костную ткань. В литературе это заболевание называют прогрессивным оссифицирующим миозитом, остеопластической миопатией, болезнью Мюншмайера или прогрессирующей оссифицирующей фибродисплазией.
Для ПОФ наиболее характерными стигматами являются как оссификация мягких тканей, так и многочисленные дефекты развития. Чаще всего имеет место микродактилия большого пальца стопы или, реже, стоп. Нередко наблюдаются короткий большой палец кисти, гипоплазия фаланг, вальгусное отклонение первых пальцев стоп, клинодактилия пятого пальца, врожденные дефекты шейного отдела позвоночника, короткие широкие шейки бедренных костей halluх valgus. Возможны глухота, облысение волосистой части головы, нерезко выраженная задержка психического развития, гипогонадизм, аномалии развития мочек уха [2]. В литературе имеются указания на ассоциацию этого заболевания с другими дефектами развития, в частности с искривлением V пальца обеих кистей, наличием 11 грудных позвонков и соответственно 11 пар ребер. Наличие таких признаков, даже при отсутствии клинической и рентгенологической симптоматики поражения мягких тканей, позволяет диагностировать это заболевание [3].
ПОФ чаще всего начинается в первой декаде жизни, но возможен дебют в более позднем возрасте или сразу после рождения [4, 5]. Как правило, первым симптомом является развитие болезненных инфильтратов в мягких тканях, которые напоминают подкожные опухолевидные образования. Возможно развитие оссификатов и без предшествующей клинически выраженной инфильтрации. Появление инфильтратов сопровождается незначительным повышением температуры и покраснением кожи над ними. Наиболее часто они локализуются в параспинальных мышцах, мышцах плечевого и тазового пояса или на шее. Однако они могут располагаться в любом месте на туловище или конечностях. В процесс вовлекаются не только мышцы, но и другие мягкие ткани, включая апоневрозы, связки, сухожилия [6]. Образование этих инфильтратов провоцируется ушибами, оперативными вмешательствами, внутримышечными инъекциями, при этом имеют значение любого вида травмы, причем нередко они могут быть довольно легкими. При гистологическом исследовании инфильтратов определяется значительная пролиферация фибробластов, которая приводит к компрессии мышечных волокон и их дистрофии. Мононуклеарные инфильтраты располагаются в мышцах и нередко спаяны с подкожной соединительной тканью.
Через несколько недель или месяцев в области этих инфильтратов происходит отложение кальция, которое в дальнейшем заканчивается оссификацией. Гетеротопические оссификаты имеют различную локализацию и могут образовываться почти во всех тканях организма, включая скелетную мускулатуру, сухожилия, связки, суставную капсулу, жировую клетчатку. При ПОФ наиболее часто встречается оссификация шеи, позвоночника и плечевого пояса, и только позже она распространяется на другие области туловища и конечности. Неуклонное прогрессирование такой оссификации развивается в направлении от позвоночника к периферии, от головы к конечностям, от проксимальных отделов к дистальным. Помимо окостенения мышц, возможно развитие экзостозов на костях конечностей, преимущественно на метафизах длинных костей, позвоночнике, костях таза, черепа или другой локализации. Прогрессирующее течение ПОФ и выраженная оссификация ведут к тяжелым осложнениям, включая тортиколиз с вовлечением m. sternoclaidomastoideus и формированием кривошеи, деформации верхней половины грудной клетки, сколиозу и тугоподвижности суставов.
В литературе представлены описания отдельных случаев ПОФ [3, 7], хотя имеются работы, в которых авторы опираются на анализ 28–32 больных с этим заболеванием [8, 9]. R. Smith и соавт. проанализировали клиническую картину ПОФ у 28 больных, которых они наблюдали в течение 24 лет [10]. Оссификация больших скелетных мышц впервые начиналась с момента рождения и продолжалась до 16 лет включительно. Ее направленность шла с шеи и мышц верхней части спины к мышцам бедра, челюсти и мышцам вблизи крупных суставов. Скорость нарастания малоподвижности и ее выраженность не были связаны с возрастом начала болезни и первичной локализацией инфильтратов. У всех больных верификация диагноза представляла большие трудности. Правильный диагноз, как правило, ставился после появления очагов эктопической оссификации, что в среднем наблюдалось через 2,7 года от начала заболевания.
В развитии ПОФ значительное место принадлежит генетической предрасположенности. Об этом свидетельствует как высокая частота разных врожденных дефектов, достигающая 75%, так и семейная агрегация этой патологии. Многие авторы рассматривают ПОФ как аутосомно-доминантное заболевание с полной пенетрацией, но различной экспрессивностью гена [4, 11]. Хотя большинство зарегистрированных случаев ПОФ является спорадическим (единичным в семье), в литературе зафиксированы и семьи, в которых поражены родитель–потомок или потомки, братья и сестры, а также близнецы. Так, J. Connor и соавт. описали семью, в которой ПОФ была диагностирована у 4 лиц трех поколений, причем в первом поколении был поражен мужчина, который имел двух больных дочерей и одну больную внучку [5]. В этой семье наблюдался выраженный полиморфизм тяжести ПОФ. В первом поколении пораженный мужчина имел асимптоматическое течение заболевания. Первым клинически выраженным проявлением была тугоподвижность шеи и спины, которая развилась после травмы. В дальнейшем, также после травмы, появилось значительное ограничение движений нижней челюсти и конечностей. Больной мог самостоятельно ходить, передвигался с помощью палочки до 70 лет и умер в возрасте 72 лет от инфаркта миокарда. При рентгенологическом исследовании выявлены множественные оссификаты мягких тканей и прогрессирующий анкилоз шейного отдела позвоночника, а также микродактилия большого пальца стопы. У одной из его дочерей заболевание проявилось в 22 года, когда после экстракции зубов и зубного протезирования у нее развилось ограничение подвижности нижней челюсти. У ее сестры в 15-летнем возрасте спонтанно припухла левая голень. После биопсии диагностирована фибросаркома. В дальнейшем у нее появились безболезненные припухлости на спине, которые, как правило, провоцировались травмой, и ограничение подвижности суставов. Заболевание имело быстро прогрессирующее течение, что привело к полной обездвиженности больной. Она умерла в возрасте 28 лет от пневмонии. У внучки в возрасте 13 лет появились болезненные припухлости на спине, а через 10 лет был выявлен крупный оссификат в области поясницы. В дальнейшем у нее были зафиксированы множественные оссификаты в бедрах и тазобедренных суставах.
Не менее интересное наблюдение о передаче ПОФ от пораженного отца двум его дочерям и сыну представили F. Kaplan и соавт. [4]. Двадцатисемилетний отец, третий сын 33-летнего здорового отца и 38-летней здоровой матери (два брата также здоровы) заболел в раннем детстве, когда впервые были выявлены фиброзные узлы в мышцах. В 2 года у него диагностирована эктопическая оссификация мышц шеи, наступившая после травмы. Болезнь прогрессировала с появлением тяжелых аномалий. У больного имелся типичный врожденный дефект большого пальца стопы. На рентгенограммах выявлялись широкие короткие шейки бедер. Все три его потомка имели характерный дефект больших пальцев и в дальнейшем у них развилась типичная картина ПОФ. У одного из сыновей оссификация в квадрицепсе появилась в 2-месячном возрасте после внутримышечной инъекции. У двух других детей инфильтраты в мягких тканях выявлены в 3-летнем возрасте.
При поиске специфических генов, ответственных за развитие ПОФ, A. Shafrits и соавт. выявили повышенную экспрессию гена BMP4 (bone morphogenic protein 4 – ВМР4) и его матричной РНК (мРНК) в лимфобластоидных клетках у 26 из 32 больных с этим заболеванием [8]. Они же обнаружили суперэкспрессию гена BMP4 и его мРНК у мужчины, страдающего ПОФ, и трех его детей, которые имели это же заболевание, в то время как среди его здоровых потомков такой закономерности не наблюдалось.
BMP4 является чрезвычайно важным регуляторным белком, который индуцирует развитие мезодермы, формирование зубов, костеобразование и репарацию переломов. Семейство костных морфогенных белков относится к суперсемейству трансформирующих факторов роста. В настоящий момент идентифицировано 9 костных морфогенных белков – ВМР 1, 2, 3, 4, 5, 6, 7, 13, 14. Все они в той или иной мере задействованы в процессах формирования хряща и кости и являются частью Wnt-сигнального пути. Тот факт, что мутации в гене ВМР4 у больных ПОФ не выявлены, а суперэкспрессия этого гена установлена не у всех больных, заставляет предположить наличие мутаций среди генов, продукты которых взаимодействуют с ВМР4. В частности, показано, что для белка BMP4 имеются соответствующие рецепторы и кодирующие их синтез гены – BMPR1A, BMPR1B и BMPR2, мутации в которых, как полагают, также могут обусловливать возникновение ПОФ при соответствующей норме последовательности гена BMP4.
Естественным эндогенным фактором, ингибирующим экспрессию гена BMP4, является Noggin-протеин, синтез которого кодирует ген NOG. Мутации в гене NOG были выявлены у больных с проксимальным симфалангизмом, который по фенотипическим проявлениям имеет некоторое сходство с дефектами пальцев кисти при ПОФ. G. Lucotte и соавт. предполагают, что такой ген у больных ПОФ кодирует синтез укороченного белка, который неэффективно ингибирует экспрессию гена ВМР4, и как следствие этот ген будет суперэкспрессирован [12]. Такая точка зрения послужила поводом для M. Xu и соавт. изучить мутационную изменчивость гена NOG у 31 больного ПОФ, причем у 13 из них наблюдались семейные случаи заболевания и у 18 – спорадические [9]. Авторы не выявили в этом гене ни одной мутации. Об интактности гена NOG в детерминации ПОФ свидетельствовали и отрицательные значения лод-баллов при анализе сцепления. На основании полученных данных авторы делают заключение, что мутации в гене NOG не являются широко распространенным стигматом среди больных ПОФ и не оказывают значимого влияния на детерминацию этого заболевания.
Для выявления кандидатных хромосомных участков, предположительно несущих гены, обусловливающие возникновение ПОФ, G. Lucotte и соавт., а также G. Feldman и соавт. провели широкое скринирование 22 аутосомных хромосом с использованием большого числа ДНК-маркеров (порядка 300–400) и анализ сцепления в семьях с множественными случаями заболевания [13, 14]. Были выявлены два критических участка, предположительно несущих ген чувствительности к ПОФ, один из которых располагался на 17-й хромосоме, а другой – на 4-й хромосоме. На 17-й хромосоме кандидатный ген, предположительно детерминирующий ПОФ, в настоящее время не идентифицирован. По данным анализа сцепления он может быть локализован на хромосомном участке между мини-сателлитными маркерами D17S809 и D17S1838. На 4-й хромосоме максимальные значения лод-баллов были получены для мини-сателлитных маркеров D4S1625 и D4S2417, фланкирующих хромосомный участок, на котором локализован наряду с другими ген SMAD1.
В настоящее время идентифицированы 7 генов SMAD – 1, 2, 3, 4, 5, 6 и 7. Белки трансформирующего фактора роста-b (TGF-b) функционируют как передатчики сигналов с гена TGF-b на соответствующие рецепторы этого гена (TGF-bR) и с гена ВМР4 на рецепторы этого гена – BMPR. Основная функция гена TGF-b – ингибиция пролиферации различного рода клеток через рецепторы и белки SMAD. Стимуляция гена TGF-b ведет к активации генов SMAD2 и SMAD3, усиливая синтез соответствующих белков, которые образуют комплекс с белком SMAD4. Последний регулирует транскрипцию генов-мишеней TGF-b-сигнального пути, которых на данный момент идентифицировано 69. Вариабельность в нуклеотидной последовательности гена TGF-b обусловливает развитие фиброза при многих патологических состояниях, например при болезнях почек, легких, диабетической нефропатии и др. Мутации в гене TGF-b вызывают болезнь Камурати – Энгельмана (Camurati–Engelmann) – заболевание, характеризующееся прогрессирующим гиперостозом и склерозом диафизов трубчатых костей (диафизальная дисплазия).
С точки зрения этиопатогенеза ПОФ, в качестве гена-кандидата чувствительности к заболеванию небезынтересен ген эктонуклеотид пирофосфатазы (ENPP1), который продуцирует неорганический пирофосфат, являющийся главным ингибитором кальцификации и минерализации. Сигнал с гена TGF-b повышает экспрессию гена ENPP1, что способствует усиленной выработке неорганического пирофосфата и уменьшает интенсивность процессов минерализции и кальцификации. Мутации в гене ENPP1 вызывают аномальную оссификацию длинных связок спины и ювенильную идиопатическую оссификацию артерий [15, 16].
Таким образом, в настоящее время гены ПОФ не идентифицированы. Можно предположить, что это заболевание детерминируется разными генами, т. е. оно генетически гетерогенно. Однако не исключено, что ПОФ может быть следствием взаимодействия нескольких генов, действующих как синергисты.
Своевременная диагностика этого заболевания оставляет желать лучшего. Причина несвоевременного распознавания в основном связана с малой информированностью врачей об этой патологии и, в частности, недиагностированностью аномалии большого пальца стопы или других дефектов развития скелета, которые являются значимым маркером этого заболевания. Диагностика ПОФ базируется на двух клинико-рентгенологических критериях, а именно наличии гетеротопической оссификации мягких тканей и аномалий развития прежде всего большого пальца стопы/стоп. При этом заболевании нет какого-либо характерного лабораторного маркера. Возможно повышение сывороточного уровня щелочной фосфатазы, свидетельствующей об активации метаболизма костной ткани, а также повышение индекса кальций/фосфор. Другие показатели, включая белковые фракции и острофазовые белки, биохимические и иммунологические тесты, не имеют отклонений от нормальных величин.
При проведении дифференциального диагноза следует иметь большую группу заболеваний, характеризующихся идиопатическим фибровоспалительным процессом. К ним относятся фокальный cклеротический миозит, воспалительный фиброзный полип желудочно-кишечного тракта, кальцифицирующий фиброзный псевдотумор, склеротический перитонит. Наиболее трудной представляется дифференциация ПОФ и ювенильного дерматомиозита с кальцинозом. Если для ПОФ типичным является сочетание аномалий развития с характерным эктопическим формированием костной ткани на туловище, то при ювенильном дерматомиозите кальцификации подвержены мышцы верхних или, что более характерно, нижних конечностей [9]. При ПОФ возможно наличие умеренной миопатии, которая является вторичной по отношению к эктопическому формированию кости.
Приводим собственное наблюдение больного с ПОФ.
Больной З., 23 года. Инвалид I группы. Закончил 12 классов средней школы и 3 курса технического университета.
Наследственность не отягощена. Мать – русская, отец – мордвин. Родители и младший брат здоровы. Других детей в семье не было. Родился здоровым мальчиком, но в первые месяцы жизни не было бровей. С 6 мес до 1 года выявлен экссудативный диатез, который появился после какой-то прививки. В раннем детстве болел краснухой и ветрянкой. Наблюдалась анемия. Гемоглобин быстро нормализовался после лечения цианокобаламином. В 4-летнем возрасте дважды наблюдался отек лба, который расценили как последствие укуса комара или пчелы. В возрасте 6,5 года без видимой причины появилась отечность голени. Температура оставалась нормальной. Позже заметили ограничение подвижности в голеностопном суставе. При рентгенологическом исследовании выявлен экзостоз. В этот период, как и на всем протяжении заболевания, СОЭ не повышалась. Проведено оперативное вмешательство с целью удаления экзостоза. После снятия гипса развилась стойкая контрактура, а через 21 день после операции обнаружили распространение депозитов кальция в мягких тканях бедра и голени. По данным гистологического исследования биоптата из области послеоперационного рубца диагностировали оссифицирующий миозит. В дальнейшем больной повторно оперировался с иссечением оссифицирующих мышц ног, что приводило только к нарастанию гетеротопической оссификации. Развились стойкие сгибательные контрактуры, из-за которых с трудом передвигался. Дважды надрезали ахиллово сухожилие, что приводило лишь к кратковременному эффекту. Сформировалась деформация стопы в положении переразгибания. В 1992 г. упал с качелей на спину. Через 2 мес развился отек шеи, а после его разрешения появилась тугоподвижность в шейном отделе позвоночника. С 1992 г. стали появляться многочисленные опухолевидные образования на спине, достигающие 15x25 см и имеющие деревянистую плотность. В 2003 г. после травмы (упал на улице) появилась припухлость бедра с гиперемией кожи и повышением локальной температуры. Эта припухлость держалась не менее 1,5 мес. Одновременно наблюдалась припухлость и в области грудино-ключичной мышцы.
При поступлении. Общее состояние средней тяжести. Ходит только с помощью посторонних лиц. Нарушена осанка. Голова отклонена влево и приведена к туловищу. Не может опираться на всю ступню и передвигается на носочках. Укорочены большие пальцы стоп. Определяются множественные экзостозы, наиболее крупные в области локтевых суставов. В месте операционного рубца на правой голени – плотное образование в виде тяжа 1,5x8 см. На левой голени определяется плотный тяж 4x10 см. Выраженная деформация грудной клетки, преимущественно в области плечевого пояса, со значительным ограничением подвижности в плечевых суставах и приведением рук к туловищу. В правом плечевом суставе почти отсутствуют пассивные или активные движения. Значительное ограничение подвижности в локтевых и левом лучезапястном суставах, а также в тазобедренных и коленных. Сгибательные контрактуры коленных, плечевых и локтевых суставов. Крупные оссификаты на спине, преимущественно в верхних отделах, безболезненные при пальпации, достигающие в длину 22 см и более. Мышцы спины плотные на ощупь. Выраженная деформация позвоночника, сколиоз грудного и поясничного отделов, ограничение подвижности шейного отдела позвоночника в трех направлениях.
В клиническом анализе крови, мочи, при детальном биохимическом и иммунологическом исследовании отклонений от нормы не выявлено. На рентгенограммах туловища и конечностей имеется большое количество крупных оссификатов разной формы и размеров (рис. 1, а, б; рис. 2), а при рентгенографии дистальных отделов стоп обнаружена микродактилия больших пальцев стоп и их синкарпия, утолщение дистальных эпифизов I плюсневых костей (рис. 3).

Рис. 1. а. Крупный оссификат в области а) наружной поверхности коленного сустава (прямая проекция) и в области б) коленного сустава (боковая проекция)
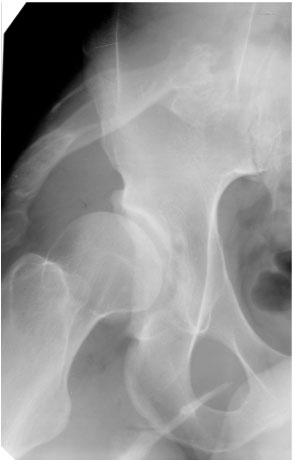
Рис. 2. Гетеротопический оссификат в мягких тканях таза
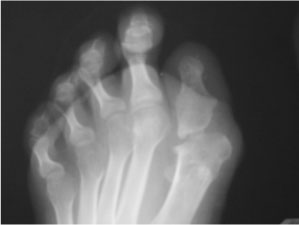
Рис. 3. Аномалия развития большого пальца стопы
В приведенном примере диагноз ПОФ основывается на наличии гетеротопической оссификации и аномалий развития (короткие большие пальцы стоп с синкарпией, утолщение дистальных эпифизов I плюснефаланговых костей, отсутствие бровей). Характерными симптомами ПОФ в данном конкретном случае являются начало заболевания в раннем возрасте, преимущественная локализация оссификаций в плечевом поясе, шее, проксимальных отделах верхней конечности, спине. Многочисленные хирургические вмешательства способствовали лишь дальнейшему прогрессированию патологического процесса. Заболевание привело к глубокой инвалидности. В стационаре назначен фосамакс, который больной принимал и после выписки из стационара. На фоне приема этого препарата (в течение 19 мес.) ни разу не появлялись воспалительные инфильтраты и не было прогрессирования гетеротопической оссификации.
Этиологическая терапия ПОФ не разработана, так как не известен причинный фактор развития этого заболевания. Терапия нестероидными противовоспалительными препаратами, иммуносупрессантами цитостатического действия, а также кортикостероидами, включая назначение высоких доз преднизолона, неэффективна. Следует отметить резистентность клинико-рентгенологических проявлений ПОФ и к другим видам медикаментозной терапии. В плане патогенетической терапии заслуживает внимание назначение интерферона-g [3]. Известно, что этот препарат уменьшает пролиферацию и метаболизм фибробластов. Длительная терапия интерфероном-g (по 800–1 000 000 ЕД 3 раза в день в течение 6 мес.) способствовала регрессу подкожных и мышечных инфильтратов. Имеются единичные сообщения о целесообразности применения бисфосфонатов [17]. Терапия бисфосфонатом (бенефос 400 мг/сут.) в течение 26 мес. способствовала существенному уменьшению частоты возникновения инфильтратов, нормализации сывороточного уровня щелочной фосфотазы и показателей обмена кальция и фосфатов, а также снижала темпы прогрессирования гетеротопической оссификации [3]. Однако место и значение бисфосфонатов в комплексной терапии ПОФ требует дальнейшего изучения. Целесообразно применение этилендиаминтетрауксусной кислоты, которая способна образовывать комплексные соединения с различными катионами и ионами кальция.
По поводу вторичной профилактики или профилактики дальнейшего прогрессирования патологического процесса можно с большой уверенностью утверждать, что следует избегать любых, даже незначительных травм, ятрогений, включая не только хирургические вмешательства, но и любые инъекции. В будущем, возможно, будут разработаны антагонисты ВМР4, причем высокую их эффективность можно ожидать не только при ПОФ, но и при ряде других идиопатических фибросклеротических состояний.
Фибродисплазия характеризуется врожденной костной патологией и прогрессирующим гетеротопическим окостенением мышц, сухожилий, связок, апоневрозов [1, 2]. Нет расовой, половой или географической предрасположенности. Заболевание чаще возникает как результат спонтанной новой мутации. Наследственная передача — аутосомно-доминантная, возможен материнский мозаицизм.
В 2006 г. была обнаружена преимущественно одна и та же гетерозиготная мутация (c.617G>A; p.R206H) в глициновом и сериновом остатке (GS) области активации рецептора 1-го типа активина А (ACVR1)/активиноподобной киназы 2 (ALK2) рецептора 1-го типа костного морфогенетического протеина (КМП), расцененная как генетическая причина ФОП. Семь дополнительных мутаций были идентифицированы впоследствии в GS-области и области киназы ACVR1 у пациентов с «нетипичными» формами болезни [3, 4].
Мутация c.617G>A приводит к замене аргинина на гистидин в кодоне 206 (p.R206H). Структурное гомологичное моделирование белков предсказывает, что замена аминокислоты приводит к конформационным изменениям рецептора, что вызывает изменение его чувствительности и активности [3].
При ФОП предполагается активная выработка КМП-4 сверх порога, что объясняет избыточную оссификацию и эктопическое образование кости постнатально [4].
Врожденные фенотипы, вызванные ФОП-метаморфозом, включают ряд врожденных уродств (деформаций) и скелетных аномалий: деформированный большой палец ноги (основной диагностический признак болезни), дисплазию метафизов, короткие фаланги, короткие большие пальцы рук, синостозы — симфалангизм пальцев, сращение поверхностей шейных суставов, сращение реберно-позвоночных суставов, проксимальные медиальные берцовые остеохондромы, короткие широкие шейки плечевой кости, редкие волосы, глухоту и др. [1, 4, 5].
В отечественной литературе имеются единичные работы о развитии ФОП у детей преимущественно в школьном возрасте [6–8].
В данном сообщении мы представляем наиболее типичные клинические и рентгенологические проявления ФОП у 30 детей в возрасте от 1,5 до 14 лет, наблюдавшихся в клинике детских болезней Первого МГМУ им. И. М. Сеченова в 1968–2010 гг. и два наблюдения с выявленным геном ФОП.
Дети поступали с жалобами на появление на голове мягких эластичных образований, которые постепенно распространялись вниз и уплотнялись. Начало болезни нередко сопровождалось недомоганием, субфебрилитетом, болезненностью очагов, постепенным их уплотнением, нарастанием общей скованности и контрактурами в области крупных суставов. Быстрота распространения процесса колебалась от 2–3 месяцев до 3–5 лет.
У отдельных пациентов наблюдали самопроизвольное исчезновение и/или рецидивы очагов, у других процесс неуклонно прогрессировал. Постепенно дети как бы окутывались костным панцирем, «вторым скелетом», становились малоподвижными, с трудом самостоятельно одевались, принимали пищу, меняли положение в пространстве и в итоге становились инвалидами, зависящими от помощи взрослых (рис. 1, 2). Дети дошкольного возраста могли отставать от сверстников в развитии, старшие были более замкнутыми, но неплохо успевали в школе.
Около половины детей имели типичный признак «классического» варианта ФОП: патологию первых пальцев стоп: укорочение, подвывихи, симфалангизм, монофалангизм и др.
На рентгенограммах обнаруживали внескелетные, костной плотности тени (оссификаты), как единичные, так и сливающиеся между собой в обширные конгломераты. Обычно определялись множественные линейные тени костной плотности с локализацией в мягких тканях конечностей и туловища. Нередко тени переплетались между собой наподобие ветвей дерева. Наиболее частой локализацией были боковые поверхности грудной клетки и внутренние поверхности плеч с образованием синостозов между собой в виде «моста» (рис. 3). У детей младшего возраста эта локализация была первичной. Нередкими были также единичные или множественные экзостозы.
Основные лабораторные параметры, включая общие и биохимические анализы крови и мочи (в т. ч. ферменты и показатели минерального обмена), уровень гормонов щитовидной и паращитовидных желез, оставались нормальными.
Лечение проводили кортикостероидными гормонами (преднизолоном или метилпреднизолоном в дозе 0,5–1 мг/кг массы в сутки) короткими курсами, нестероидными противовоспалительными препаратами (НПВП), внутривенным введением 5% раствора динатриевой соли этилендиаминтетрауксусной кислоты (Na2 ЭДТА), 10% раствора Ксидифона внутрь и/или местно путем электрофореза или мази с Ксидифоном. В последние годы использовали бисфосфонаты нового поколения (динатрия памидронат или Аредиа, Бонефос, Бонвива и др.), антилейкотриеновые препараты (монтелукаст) и блокаторы мастоцитов (кромолин натрия).
Приводим представляющие интерес наблюдения двух пациентов, обследованных генетически в 2010 году.
Артем Т. в 14 лет обратился в клинику в июне 2010 года.
Беременность у матери протекала нормально, но в конце появились отеки ног, А/Д — 150/100 мм рт. ст, прибавка веса 10 кг. Из-за запрокидывания головки ребенка проведено кесарево сечение (12-часовой безводный период). Вес при рождении 3200 граммов, длина 53 см, окружность головки 36 см. Рос и развивался удовлетворительно. Сидит с 6 месяцев, начал ходить с 10 месяцев, первый зуб прорезался в 4 месяца. Вес в один год 11 кг, речь с 1 года 3 месяцев. Грудное вскармливание 2 месяца, далее искусственное, диагностирована анемия легкой степени.
На все прививки, кроме реакции Манту, отмечали подъем температуры до 39–40 °C. При проведении АКДС распухала ягодица (как «стеклянная»).
Перенесенные болезни до школы — ветряная оспа, мононуклеоз, позже — ОРВИ.
В два года поставлен диагноз врожденная вальгусная деформация больших пальцев стоп, сделана операция выправления подвывиха первого пальца левой ноги (вставлена спица).
В 5 лет — диагноз болезнь Пертеса, экзостозная болезнь.
В 6 лет сделана операция по удалению «наростов» (экзостозов) с обеих сторон на задней поверхности коленных суставов. Но возник рецидив, и появились новые очаги спереди.
В 9 лет из-за ушиба (падение с велосипеда) стала плохо сгибаться в колене левая нога, стал прихрамывать.
С 11 лет выявлена двухсторонняя нейросенсорная тугоухость.
В 14 лет (апрель 2010 г.) появилось уплотнение на шее слева. На операции в кивательной мышце выявлен тяж хрящевой плотности. Материал биопсии консультирован в РОНЦ АМН РФ: данных за опухоль нет.
Консультирован в ЦИТО в мае 2010 г., установлены экзостозы и ограничение движений в левой кивательной мышце.
УЗИ левой кивательной мышцы (21.05.2010) — в средних ее отделах определяется участок измененной структуры, захватывающий всю толщу мышцы на протяжении 5,2 см с уплотнением, понижением эхогенности, не имеющий характерной волокнистой структуры; выше и ниже в мышечной ткани есть участки гиперэхогенных мышечных включений (послеоперационные изменения на фоне специфического миозита). Справа — аналогичные включения.
УЗИ передней поверхности правого бедра — на уровне верхней трети — оссификат (2,8 × 0,3 см толщины), в мягких тканях спины справа, на уровне нижних грудных позвонков — оссификат, аналогичный по структуре.
По завершении обследования и очной консультации в РОНЦ им. Н. Н. Блохина в мае 2010 года заподозрен прогрессирующий оссифицирующий миозит.
На консультации генетика установлен диагноз — «фибродисплазия прогрессирующая оссифицирующая с аутосомно-доминантным типом наследования в родословной «de novo». В фенотипе — уплотнение мышц в области шеи, спины, правого бедра, с 9 лет костно-хрящевые экзостозы в верхней трети большеберцовой кости и нижней трети левого бедра, врожденная аномалия первых пальцев кистей и стоп (укорочение, тугоподвижность в межфаланговых суставах), укорочение и расширение шейки бедренных костей.
В клинике детских болезней обследован амбулаторно (15–17.06.2010 г.).
При осмотре мальчик хорошего роста, повышенного питания (рост 160 см, вес 60 кг). Жалуется на затруднение при поворотах шеи и наклонах туловища вперед. Ощущает неловкость в спине и тазе при ходьбе и ощупывании, прихрамывает на правую ногу.
Кожа чистая со следами загара. Укорочение больших пальцев стоп и кистей с подвывихом на ногах — halux valgus (рис. 4). На шее в области левой кивательной мышцы уплотнение с рубцом в центре. Поворот влево затруднен. Наклон туловища вперед лишь до горизонтального уровня. Справа на передней поверхности бедра — уплотнение в толще мышцы. На спине справа на уровне 6-го грудного позвонка определяется костный тяж длиной около 5–7 см, вне связи с позвоночником. Лимфатические узлы не увеличены. Со стороны легких, сердца, органов брюшной полости без патологии.
На R-грамме стоп: справа — вальгусная деформация первого пальца, основная фаланга значительно укорочена, утолщена, смещена латерально (подвывих). Слева — деформация головки плюсневой кости и проксимальной фаланги первого пальца (после операции). Суставная щель неравномерно сужена (рис. 4).
На рентгенограмме тазобедренных суставов с захватом 2/3 бедренных костей — головки бедренных костей уплощены, левая смещена латерально. Слева крыша вертлужной впадины скошена. Утолщение кортикального слоя в бедренных костях. В проекции шейки и верхней половины диафиза бедренной кости слева в мягких тканях определяются тени костной плотности. Справа — единичные тени в проекции шейки бедренной кости (рис. 5).
На рентгенограммах кистей соотношение костей не нарушено. Отмечается метаэпифизарный остеопороз. Пястные кости укорочены. Слева основная фаланга первого пальца укорочена, гипоплазия эпифиза. Средние фаланги V пальцев с искривленностью.
ЭхоКГ (17.06.2010 г). Размеры полостей сердца, толщина миокарда в пределах нормы. Систолическая и диастолическая функции левого желудочка не нарушены. Давление в легочной артерии в норме. По передней стенке правого желудочка минимальное расхождение листков перикарда в диастолу 1,3–1,5 мм. МАРС: добавочные базальные хорды и трабекула в полости левого желудочка. Удлинен евстахиев клапан.
Клинический диагноз: «Фибродисплазия оссифицирующая прогрессирующая. Классическая форма. Поздняя стадия. Врожденные аномалии больших пальцев стоп: укорочение и утолщение основных фаланг с подвывихом (Halux valgus) и кистей (укорочение пястных костей и основной фаланги первого пальца слева с гипоплазией эпифиза. Искривление средних фаланг V пальцев). Костно-хрящевые экзостозы слева на уровне нижней и верхней третей бедренной кости. Укорочение и расширение шейки бедренных костей. Множественные внескелетные костные образования (оссификаты) в толще мышц спины, бедер, шеи.
МАРС: добавочные базальные хорды и трабекула в полости левого желудочка, удлинение евстахиевого клапана. Двусторонняя нейросенсорная тугоухость.
В лаборатории ДНК-диагностики: найдена мутация Arg206 His в гене ACVR1».
Заключение. Ребенок наблюдался длительно с диагнозами врожденной вальгусной деформации больших пальцев стоп, экзостозной болезни, болезни Пертеса, подозрением на опухоль шеи с биопсией измененного участка кивательной мышцы; получал все прививки и вакцинации, несмотря на высокую лихорадку на каждое введение вакцин, не соблюдал необходимого щадящего двигательного режима, многократно травмировался, подвергался многочисленным лучевым исследованиям, операциям и лишь через 14 лет диагноз ФОП верифицирован и подтвержден генетически.
В настоящее время ему рекомендованы щадящий двигательный режим, отвод от всех внутримышечных инъекций, манипуляций в области нижней челюсти, профилактика ОРВИ (Арбидол, санация полости носа — промывание морской водой, капли — Маример, вакцинация при эпидемии гриппа подкожно или интраназально), плавание в бассейне с морской водой или в море, применение преднизолона (2 мг/кг 5–7 дней) и бисфосфонатов (Аредиа) при возникновении новых оссификатов, оформление инвалидности и динамическое наблюдение в специализированном стационаре.
Второй пациент — Алеша О. под наблюдением клиники с 1 года 3 месяцев.
В анамнезе: мать 27 лет, здорова, отец 39 лет, страдает бронхиальной астмой, алкогольной зависимостью. На 18-й неделе беременности мать перенесла грипп, в последующем из-за непропорционального развития головки была госпитализирована в отделение патологии беременности. Роды в срок, физиологические. Вес ребенка 2530 г, длина — 48 см. Оценка по шкале Апгар 8–9. Закричал сразу. Кормление грудью 8 месяцев, отмечалась гипогалактия. С раннего детства отмечено «плохое» держание головки в положении на животе, в 3 месяца — массаж из-за гипертонуса мышц шеи. Сидит с 7 месяцев, ходит с 12 месяцев, зубы с 5 месяцев.
В возрасте 5 месяцев упал с дивана без последствий. Позже в 12 месяцев повторное падение, ударился лбом. На месте ушиба возник отек размером с куриное яйцо. Получал гидрокортизон, примочки местно с эффектом.
При рентгенографии и томографии выявлена врожденная аномалия С1–С2 — деформация тел позвонков в виде платиспондилии, незаращение на уровне задних отделов (spina difida posterior), гипоплазия зубовидного отростка С2.
При ЭхоКГ размеры камер сердца выше возрастной нормы (мальчик повышенного питания: вес в 1 год 2 месяца 15 кг). Дополнительная хорда в левом желудочке.
При первичном осмотре ребенок хорошего физического развития, повышенного питания, мобилен, общителен. Имеется «скованность» и приподнятость в плечевом поясе, напряженность и уплотнение мышц шеи, ограничение в поворотах головы и наклоне назад. Движения в суставах конечностей не ограничены, деформаций нет. Приседает, ходит, бегает свободно. Кожные покровы и слизистые чистые. Зубы 10/8. По внутренним органам — без патологии, выявлена гипоспадия.
Имеются диагностические признаки классической формы ФОП — укороченные с подвывихом большие пальцы стоп (рис. 6), а также врожденная патология шейных позвонков и гипоспадия.
Общие анализы крови и мочи, биохимическое исследование крови в пределах нормы.
Генетическое обследование выявило наличие мутации Arg206His в гене ACVR1, характерное для классического варианта ФОП.
Клинический диагноз: «Фибродисплазия оссифицирующая прогрессирующая. Классическая форма. Ранняя стадия. Начальная фаза болезни. Рецидивирующие опухолеподобные образования на голове и в области шеи. Хрящеподобные элементы в виде продольных эластичных тяжей (теней на рентгенограммах) в мягких тканях надплечий и вдоль позвоночника. Врожденные костные аномалии: укорочение больших пальцев стоп. Недоразвитие зубовидного отростка 2-го шейного позвонка. Платиспондилия и незаращение задних отделов тел С1–С2. Гипоспадия. Мутация Arg206 His в гене ACVR1».
Ребенок наблюдается в течение года. Заболевание не прогрессирует (рис. 7).
Периодически на голове при легких травмах возникают эластичные «опухоли», быстро исчезающие при применении мази с Ксидифоном. Дан отвод от всех внутримышечных прививок и вакцинаций. Рекомендован охранительный двигательный домашний режим. При падениях и образовании мягких опухолей на голове — мазь с Ксидифоном, внутрь НПВП, антилейкотриены. Динамическое наблюдение в клинике для коррекции терапии. Оформление инвалидности детства.
Таким образом, наши наблюдения свидетельствуют о недостаточном знакомстве с ФОП врачей различных специальностей — педиатров, хирургов, ортопедов, что приводит к длительной верификации диагноза, ненужным вмешательствам и травмам мягких тканей (операции, биопсии, внутримышечное введение лекарств, вакцинации).
Обобщая литературные и собственные данные, следует считать для диагноза главным наличие врожденной патологии больших пальцев стоп (нередко в сочетании с патологией пальцев кистей) и внескелетные оссификаты. У детей дошкольного возраста, как правило, они возникают на шее, плечевом поясе и постепенно распространяются вниз. У старших детей оссификаты могут появляться в любых участках мягких тканей (мышцах, сухожилиях и др.), чаще вследствие травм любого генеза или на фоне гриппа и ОРВИ. При подозрении на ФОП необходим поиск мутаций в гене ACVR1.
К сожалению, прогноз болезни неудовлетворительный. Очень важно соблюдать все профилактические меры для избежания любых травм мягких тканей и проводить профилактику ОРВИ и гриппа, которые могут ускорить прогрессирование болезни и спровоцировать легочно-сердечную недостаточность.
В настоящее время содружеством ученых активно разрабатываются и апробируются (в эксперименте и на животных) препараты, обладающие способностью блокировать мутации в гене ACVR1, что позволит предупредить или прервать гетерогенную оссификацию и улучшить состояние пациентов.