Современные подходы к классификации аномалий мозжечка (обзор литературы). Клеймёнова И.С.
Интенсивное развитие современных методов нейровизуализации сопровождается выявлением множества пороков развития большого мозга и мозжечка. В зарубежной и отечественной литературе представлены как описания часто встречающихся пороков развития (Dandy-Walker continuum), так и других, реже диагностирующихся аномалий развития мозжечка (Joubert syndrome (Joubert M.,1969; Boltshauser E., 1981), Arima syndrome (Arima M., 1971), Senior-Lokensyndrome (Satran D., 1999, Loken A.C., 1961; Senior B, 1961), rhombencephalosynapsis (Truwit C.L., 1991)). Описаны, однако, недостаточно понятны причины и следствия изолированных нарушения развития червя и гемисфер мозжечка. Не существует четких критериев для проведения дифференциального диагноза между атрофией, гипоплазией и аномалией развития мозжечка (Ramaekers V.T., 2000; Ramaekers V.T., 1997; Steinlin M., 1998; Esscher E., 1996). Таким образом, прогноз развития пациентов с многочисленными церебеллярными мальформациями остается до настоящего времени не ясным.
Классификация аномалий развития мозжечка предложена Patel S et Barkovich AJ (2002). Она разработана авторами на основании анализа базы данных, насчитывающей 70 собственных наблюдений, охватывающих широкий спектр нарушений формообразования структуры. Учитывая детальный анализ вариантов возможных церебеллярных мальформаций, мы сочли необходимым подробно остановиться на результатах данного исследования.
В исследование не были включены пациенты с Chiari malformations, поскольку данный порок развития связан с нарушением образования IV эмбрионального желудочка, вследствие чего формируется гипопластичная заднечерепная ямка, и является пороком развития краниовертебральной области, а не структур мозжечка (McLone DG, 1998; StovnerLJ, 1993; Vega A, 1990). По тем же причинам в исследование не были включены пациенты с затылочным энцефалоцеле (Chapman PH, 1989). Кроме того, в исследование не были включены пациенты с изолированной гипоплазией нижних отделов червя мозжечка, поскольку данный вариант не всегда рассматривается как аномалия (Patel S., Barkovich A.J., 2003).
Прежде всего, исследователями выделены два класса пороков развития: гипоплазия мозжечка как результат недоразвития органа, проявляющийся врождённым дефицитом массы и уменьшением размера органа, и дисплазия, аномалия структур тканей и органа, как следствие нарушения гистогенеза и формообразования.
Гипоплазия мозжечка может быть следствием преждевременной остановки миграции клеток или чрезмерного апоптоза в период развития структуры. Термин применяется для описания маленького мозжечка с нормальными размерами борозд полушарий и червя мозжечка. Она может быть как генерализованной, так и фокальной, например как следствия нарушения пролиферации клеток Purkinje с сопутствующей редукцией гранулярного слоя.
Дисплазия рассматривается как результат аномальной миграции нервных клеток и нарушения организации коры мозжечка с искажением складчатости и формирования борозд (Chen S, 1989; Herrup, 1997). МР-признаком дисплазии коры полушарий мозжечка является изменение направления и размеров борозд (вертикализация борозд), патологический пограничный паттерн между серым и белым веществом полушарий мозжечка, отсутствие нормальной арборизации белого вещества, гетеротопия серого вещества или ядер мозжечка, патологический паттерн «складчатости» коры. Также к радиологическим признакам дисплазии относится истончение коркового слоя, гипертрофия и кистоподобное изменение структуры коры гемисфер мозжечка.
Термин атрофия используется для описания маленького мозжечка со «сморщенным» червём, расширенными бороздами и уменьшением объёма структуры в динамике. Атрофия мозжечка представляет собой прогрессирующее повреждение, обусловленное нарушением метаболизма, в ряде случаев сочетающееся с аномалией анатомической структуры.
В каждом классе выделяются две группы: фокальные и генерализованные нарушения развития.
Таблица 1. Классификация аномалий мозжечка (Patel S, Barkovich AJ, 2002)
I. Гипоплазия мозжечка
II. Дисплазия мозжечка
A.Фокальная гипоплазия
B.Генерализованнаягипоплазия
A. Фокальная дисплазия
B. Генерализованная дисплазия
1.Изолированная гипоплазия червя
1. С расширением IVжелудочка, комплексDandy-Walker
1. Изолированная дисплазия червя
1. Врожденная мышеч-ная дистрофия
2.Изолированная гипоплазия одной геми-сферы
2. С нормальным IVжелудочком
a. Аномалия типа «коренного зуба», ассоциированная с дисплазией ствола мозга (JoubertSyndrome)
2.Цитомегаловирусная инфекция
a. с нормальным мостом
b. Ромбэнцефалосинапсис
3.Лиссэнцефалия с мутацией RELN
b. с гипоплазией моста
2. Изолированная дисплазия гемисфер
4.Лиссэнцефалия с агенезией мозолистого тела и церебеллярной дисплазией
с. с нормальным строением из-вилин полушарий мозжечка
a.Фокальная мозжечковая кортикальная дисплазия/гетеротопия
5.Ассоциированная с диффузной церебральной полимикрогирией
a) Понтоцеребеллярная гипоплазия Барта, тип I и II
b. Синдром Лермитта-Дуклоса–Коудена
6. Диффузная аномалия извилин
b) церебеллярная гипоплазия без определённой специфичности
В 1987 году Macchi and Bentivoglio предложена теория латерального вовлечения, согласно которой фокальные аномалии развития рассматриваются как результат локального повреждения, в отличии от генерализованных аномалий, как результата генетических или метаболических нарушений. Исследование Patel S, Barkovich A.J. (2002) выявило закономерности ассоциации генерализованных мальформаций мозжечка с аномалиями большого мозга, в отличие от фокальных аномалий, при которых полушария большого мозга остаются нормальными. Анализ изображений, полученных при проведении МРТ исследования головного мозга у пациентов с диффузной дисплазией мозжечка (мышечная дистрофия (n=10), врождённая цитомегаловирусная инфекция (n=6), лиссэнцефалия (n=3)), позволил установить, что во всех случаях диффузная дисплазия мозжечка сочеталась с пороками развития большого мозга. Пациенты с фокальными формами дисплазии мозжечка (Joubert syndrome (n=12) и rhombencephalosynapsis(n=8)) имели вариабельное развитие структур большого мозга. При этом несиндромные формы фокальной дисплазии и гипоплазии мозжечка (изолированная фокальная мозжечковая кортикальная дисплазия (n=2), мозжечковая гетеротопия с церебеллярной кортикальной дисплазией (n=1), идиопатическая диффузная церебеллярная дисплазия (n=1),Lhermitte-Duclos syndrome (n=1), изолированная мозжечковая гипоплазия (n=6), понтоцеребеллярная гипоплазия тип 1 (n=1)) имели нормальный большой мозг. Пациенты с Dandy–Walker continuum (n=19) имели одновременно гипоплазию и дисплазию структур мозжечка. При этом значительных различий меду типичной Dandy–Walker malformation (наличие кисты IV желудочка, расширение ретроцеребеллярного пространства) и Dandy–-Walker variant (изолированной гипоплазии червя мозжечка с расширением IV желудочка) выявлено не было.
I. Гипоплазия мозжечка
Генерализованная гипоплазия с расширением IV желудочка (Dandy-Walker Continuum).
Аномалия структур задней черепной ямки была впервые описана в 1914 г. американским хирургом Walter E. Dandy (1886–1946 гг) и американским врачом Kenneth D. Blackfan (1883–1941 гг). В 1954 году Benda предложил термин «аномалия Денди–Уокера».
Рис 1. Dandy-Walker Malformation.
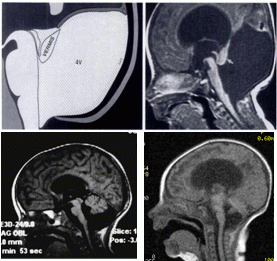
- характерная триада при Dandy-Walker Malformation: гипоплазия червя мозжечка (vermis), кистообразное расширение IV желудочка (4V), расширенная заднечерепная ямка с высоко расположенным диспластичным намётом мозжечка, венозными синусами (Spyros S. Kollias et al, 1993).
- сагиттальное T1-weighted изображение, демонстрирующее гипопластичный червь мозжечка (стрелка), приподнятый вверх расширенным IV желудочком (Spyros S. Kollias et al, 1993).
- сагиттальное T1-weighted изображение, демонстрирующее гипопластичный червь мозжечка, приподнятый вверх расширенным IV желудочком, расширение задней черепной ямки с приподнятым диспластичным намётом мозжечка (материалы НТЦ ПНИ, (Скворцов И.А., 2009).
- сагиттальное T1-weighted изображение, демонстрирующее гипопластичный червь мозжечка, приподнятый вверх расширенным IV желудочком, гидро-цефальное расширение боковых желудочков (собственные материалы, Клеймёнова И.С., 2010).
D’Agostino (1963) и Hart et al (1972) выделили характерную триаду аномалии Денди–Уокера (рис. 1):
- полная или частичная агенезия червя мозжечка,
- кистозная дилатация IV желудочка,
- расширение задней черепной ямки со смещением вверх латеральных синусов, намета мозжечка и стока синусов.
Как правило, у пациентов диагностируется супратенториальная гидроцефалия, которую следует рассматривать как осложнение, а не часть аномалии.
Таким образом, аномалия Денди–Уокера (DWM) представляет собой врожденный порок развития крыши IV желудочка и червя мозжечка, ведущий к неполному раскрытию срединной (Мажанди) и латеральной (Лушки) апертур IVжелудочка. Причиной мальформации является персистенция передней мембранозной области с её расширением и грыжевидным выпячиванием между латеральными зачатками червя мозжечка и сосудистого сплетения. Согласно этой же теории крыша III желудочка поднимается вверх, нарушая формирование мозолистого тела. Патологические факторы, ведущие к таким нарушениям должны воздействовать на развивающийся мозг в период 7–10 недель гестации. Заболевание проявляется признаками гидроцефалии, нередко гидромелии.
Радиологическими признаками Dandy–Walker Malformation является увеличение размеров черепа с характерной истончённой выступающей затылочной частью, расширение IV желудочка, высокое расположение намёта мозжечка, венозных синусов, стока синусов и расширение задней черепной ямки. Латеральные синусы оказываются смещенными вверх и при слиянии с сагиттальным синусом образуют инвертированный знак Y. Отсутствует серп мозжечка. Гипоплазия червя мозжечка лучше определяется при проведении срединного сагиттального среза. Полушария мозжечка могут быть гипопластичными.
Классическая аномалия Денди–Уокера встречается с частотой 1 на 25000–30000 новорожденных. Ohaegbulam SC, AfifiH. (2001) в течение 11 лет изучавшие частоту встречаемости синдрома в семьях военного персонала Саудовской Аравии и исследовавшие 45 274 новорожденных, оценивают её как 1 на 100 000 живорожденных, при этом частота встречаемости синдрома у младенцев мужского пола составила 1,24 на 100 000, у младенцев женского пола - 0,78 на 100 000 живорожденных.
DWM составляет 1–4% всех случаев гидроцефалии. Декомпенсированная гидроцефалия диагностируется у 75% трехмесячных младенцев с аномалией Денди–Уокера и у 90% пациентов на момент постановки диагноза. OhaegbulamS.C., Afifi H. (2001) выявили её у 3,5% младенцев с врожденной гидроцефалией. Unsgaard G. et al (1987), Sato K. et al(1996) приводят случаи собственных наблюдений и тринадцать описаний литературных источников когда первичная декомпенсация гидроцефалии происходила у взрослых людей, что позволило впервые диагностировать у них аномалию Денди–Уокера. Freeman SR, Jones PH. (2002) описывают случай первичной постановки диагноза DWM при проведении МРТ-исследования пациенту в возрасте 75 лет, который обратился к врачу впервые в связи с развитием сенсоневральной тугоухости и эпизодов головокружения. Yamamoto Y et Waga S (1984) представляют случай случайного выявления порока (DWV) у пациента 50-и лет не предъявлявшего ни каких жалоб и не имевшего никаких патологических неврологических симптомов. При проведении МРТ мозга у мужчины случайно была выявлена умеренная сообщающаяся гидроцефалия и гипоплазия нижних отделов червя мозжечка без кистозного расширения задней черепной ямки.
Raybaud при описании менее выраженной дилатации заднечерепной ямки, расширения IV желудочка, имеющей один или несколько путей оттока и гипоплазию червя мозжечка использовал термин «вариант Денди–Уокера» (DWV).
Ассоциация DWV с рядом супратенториальных мальформаций (агенезия мозолистого тела, киста диэнцефальной области, гетеротопии, аномалии строения извилин, голопрозэнцефалия, затылочное менингоэнцефалоцеле) подчёркивает одновременное формирование вышеописанных структур в нейроонтогенезе.
Spyros S. Kollias et al (1993) предложили термин вермиан-церебеллярная гипоплазия для описания мальформаций включающих неоцеребеллярную аплазию или гипоплазию ассоциированную с гипоплазией червя мозжечка и указывают на то, что применение этого термина позволит акцентировать внимание именно на аномалии развития мозга. Эта группа мальформаций отличается от DWM степенью расширения задней черепной ямки с нормальным расположением намёта мозжечка, IV желудочек лучше сформирован и менее расширен.
Рис 2. Dandy-Walker Variant.
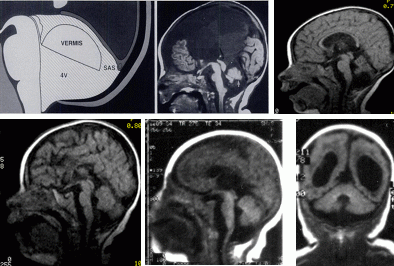
- диаграмма изображает структуры задней черепной ямки при гипоплазии червя мозжечка (DWV) (Spyros S.Kollias et al, 1993).
- сагиттальное T1-weighted изображение гипоплазии червя (стрелка) с его поворотом вверх и вперёд, киста задней черепной ямки при нормальных её размерах, ассоциированные с врожденными супратенториальными аномалиями: агенезия мозолистого тела, срединная межполушарная киста (Spyros S. Kollias et al, 1993).
- сагиттальное T1-weighted изображение гипоплазии червя с нормальным мостом, киста задней черепной ямки при нормальных её размерах ассоциированные с гипоплазией мозолистого тела (собственные данные Клеймёнова И.С., 2010)
- сагиттальное T1-weighted изображение гипоплазии червя с нормальным мостом, киста задней черепной ямки при нормальных её размерах ассоциированные с агенезией мозолистого тела (собственные данные Клеймёнова И.С., 2010).
- а) фронтальное и б) сагиттальное T1-weighted изображения гипоплазии нижних отделов червя, умеренной гипоплазии моста, расширения IV желудочка, кисты задней черепной ямки при нормальных её размерах ассоциированные с семилобарной голопрозэнцефалией (собственные данные Клеймёнова И.С., 2010).
- а) фронтальное и б) сагиттальное T1-weighted изображения гипоплазии червя с нормальным мостом, киста задней черепной ямки при нормальных её размерах ассоциированные с лиссэнцефалией, агенезией мозолистого тела, вентрикуломегалией, гипоплазией полушарий мозжечка (собственные данные, Клеймёнова И.С., 2010).
Barkovich AJ (1989) указывает на то, что аномалия Денди–Уокера, вариант Денди–Уокера, а также дилатация большой затылочной цистерны (mega cisterna magna) представляют собой нарушения одного и того же плана и объединяет их под термином «комплекс Денди–Уокера» (Dandy-Walker Continuum – DWC).
Mega Cisterna Magna порок характеризуется расширением Cisterna Magna, свободно сообщающейся с перимедуллярным субарахноидальным пространством, желудочковой системой, морфологически интактными червём и полушариями мозжечка. Cisterna Magna формируется совместно с мягкой мозговой оболочкой к концу 7 недели гестации. Анатомически располагается в углублении между поверхностью червя мозжечка и миндалин, распространяясь в затылочное пространство и кпереди до отверстия Мажанди. Позади Cisterna Magna ограничена арахноидальной мембраной, протянувшейся от верхней цервикальной хорды до задненижней поверхности мозжечка на уровне пирамид и отделяющей Cisterna Magna от цистерны, расположенной под червём мозжечка. Термин MegaCisterna Magna был предложен Gonsette et al в 1962 году. Harwood–Nash для описания подобной аномалии использовал термин Blake’s pouch.
Проведению дифференциального диагноза между компрессией и гипоплазией червя мозжечка помогает идентификация 9 нормальных долек червя, нормального ствола мозга, кроме того, в случае компрессии наблюдается передневерхнее смещение червя мозжечка.
Ряд авторов (Tortori–Donati) рассматривают как независимый синдром в пределах комплекса Денди-Уокераперсистирующую кисту Блейка (Blake’s pouch cyst –BPC). Calabro F et al (2000) описывают BPC как баллонообразное образование расположенное позади верхнего мозгового паруса в cisterna magna и представляют два случая BPC, обнаруженного в взрослых женщинах подвергшихся нейрорадиологическому исследованию из-за головной боли и эпизодов потери сознания. Результаты МРТ включили гидроцефальное расширение IV желудочка, его широкое сообщение с кистой задней черепной ямки (то есть, BPC), гипоплазию червя мозжечка и мозжечковых полушарий и отсутствие связи между четвертым желудочком и субарахноидальным пространством посредством отверстия Мажанди. BPC обусловлен внутриутробным нарушением формирования отверстия Мажанди в задней мембранозной области. В ряде случаев нормальная функция отверстий Лушки может частично обеспечивать отток цереброспинальной жидкости из желудочков в субарахноидальные пространства, однако неизбежно компенсаторное расширение мозговой желудочковой системы мозга, то есть, гидроцефалия.
Рис 3. Mega Cisterna Magna.
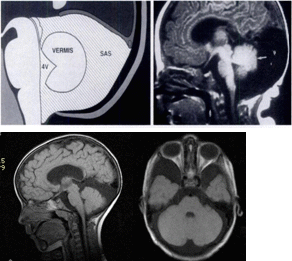
- иллюстрация Mega Cisterna Magna. 4V – IV желудочек, SAS - супраарахноидальное пространство (Spyros S. Kollias et al, 1993).
- сагиттальное T1-weighted изображение кистоподобного образования, расположенного позади нормально сформированного червя мозжечка (стрелка) изоинтенсивное сигналу ликвора. Увеличены размеры задней черепной ямки (Spyros S. Kollias et al, 1993).
- сагиттальное T1-weighted изображение кистоподобного образования, расположенного позади нормально сформированного червя мозжечка изоинтенсивное сигналу ликвора, увеличены размеры задней черепной ямки (собственные данные Клеймёнова И.С., 2010).
- аксиальное T1-weighted изображение кистоподобного образования, расположенного позади нормально сформированных полушарий и червя мозжечка изоинтенсивное сигналу ликвора, увеличены размеры задней черепной ямки (собственные данные Клеймёнова И.С., 2010).
Все варианты DWC представляют собой аномалии развития мозгового паруса, червя мозжечка и его полушарий, сосудистого сплетения четвертого желудочка, субарахноидальной цистерны задней черепной ямки и менингеальных оболочек. Tortori–Donati P. et al (1996) классифицируют кистозные пороки развития задней черепной ямки составляющие DWC на основе их происхождения и выделяют две группы: аномалии формирующиеся из передней и задней мембранозной области. Авторы приводят детальное описание различных стадий формирования структур задней черепной ямки и ее содержимого в онтогенезе и предлагают идентификацию двух аномалий, производных от дефекта задней мембранозной области: mega cisterna magna (MCM) и персистирующей кисты Блейка (BPC). Подчеркивается важность дифференциального диагноза ввиду радикально различных терапевтических подходов.
Арахноидальная киста (Arachnoid Cyst) задней черепной ямки – доброкачественное образование, формирующееся из арахноидальной мембраны, не имеющее свободного сообщения с желудочковой системой и субарахноидальным пространством. Существует две теории формирования арахноидальной кисты. Согласно первой киста формируется как дивертикул первичной мозговой оболочки, формирующейся из мезенхимы, согласно второй, киста – результат патологического развития нижней мембранозной области. Плотность ликвора в кисте приблизительно идентична плотности цереброспинальной жидкости. Кроме того, по мере роста кисты нарастает компрессия нормально сформированных полушарий, червя мозжечка и ствола мозга, что требует радикальной хирургической тактики лечения (Barkovich AJ et al., 1989). Позиция сосудистых сплетений в IV желудочке может служить подсказкой при определении этиологии кисты задней черепной ямки. Расположение этих сплетений соответствует норме в случае арахноидальной кисты, сплетения отсутствуют при Dandy Walker malformation, диспластичные и смещены вверх в случае кисты Blake(Nelson MD Jr et al., 2004).
Порок развития может быть вызван различными генетическими и средовыми факторами (Andreea Nissenkorn et al, 2001). Развитию порока способствуют токсикоз в первом триместре беременности, инфицирование вирусами краснухи, цитомегалии, токсоплазмоза, прием варфарина, алкоголя, свинцовая интоксикация. Хроническая гипоксия в первом триместре беременности провоцирует мозжечковые дисплазии у животных (Soto–Ares Gustavo et al, 2002). Agnes Messerschmidt et all (2005) указывают, что глубокая недоношенность является фактором, ведущим к дизрупции развивающегося мозжечка и нарушение развития структур задней черепной ямки вследствие недоношенности является гораздо более распространенной патологией, чем считалось до настоящего времени.
Рис 4. Arachnoid Cyst of the Posterior Fossa.
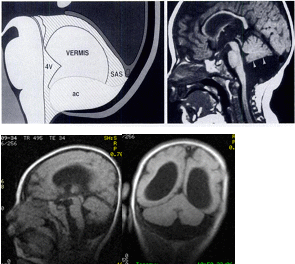
- диаграмма иллюстрирует кисту задней черепной ямки. 4V – IV желудочек, SAS - супраарахноидальное пространство, ac – арахноидальная киста (Spyros S. Kollias et al, 1993).
- сагиттальное T1-weighted изображение кисты задней черепной ямки с компрессией нормально сформированного червя мозжечка (стрелка) (Spyros S. Kollias et al, 1993).
- а) сагиттальное и б) фронтальное T1-weighted изображение кисты задней черепной ямки с компрессией червя мозжечка, гипоплазия левого полушария мозжечка (собственные данные Клеймёнова И.С., 2010).
Одной из причин формирования аномалий мозжечка являются врожденные нарушения метаболизма. Пероксисомные болезни, дефекты окисления жирных кислот ведут к нарушению миграции нейронов. Дефицит пируватдегидразы, некетотическая гиперглицинэмия и материнская фенилкетонурия являются причиной формирования пороков сочетанных с дисгенезией мозолистого тела. Нарушение обмена фолиевой кислоты ведет к дефектам невральной трубки, нарушение обмена холестерина является причиной пороков мозжечка, сочетанных с голопрозэнцефалией. Синдром врождённого нарушения гликозилирования (Steinlin M et al, 1998; Ramaekers VT et al.,1997) ведёт к уменьшению размеров мозжечка при сохранении его формы при выраженной потере белого и серого вещества. Пороки развития нервной трубки, включающие аномалию Денди-Уокера, встречаются у младенцев рожденных от женщин получавших препараты вальпроевой кислоты (Lindhout D et al, 1992).
Клинические проявления аномалии Денди–Уокера включают задержку психомоторного развития, макроцефалию, гидроцефалию различной степени выраженности (Niesen CE et al, 2002). Степень задержки психомоторного развития и дальнейший прогноз зависят от наличия сопутствующих аномалий ЦНС и внутренних органов. При изолированной DWM, единственным признаком может быть патологический прирост окружности головы. Макроцефалия обычно является следствием гидроцефалии, в некоторых случаях она связана с расширением задней черепной ямки, при этом череп приобретает характерную долихоцефалическую форму с выступающими затылочными буграми. Для большинства детей характерны атактический синдром, мышечная гипотония или спастичность, нарушение формирования мелкой моторики (Gupta PK et al, 2000). Имеются данные о дисфункции дыхательного центра ствола мозга, что может провоцировать приступы апноэ у 15-30 % пациентов.
Киста паутинной оболочки задней черепной ямки, относящаяся к DWC, обычно остаётся бессимптомной или приводит к появлению неспецифических общемозговых симптомов.
Сочетание сирингомиелии с вариантом Денди–Уокера описано в 17 публикациях (Hammond CJ, Chitnavis B et al, 2003), указывается на роль обструкции большого затылочного отверстия в нарушении ликвородинамики на уровне ствола и спинного мозга. Harriette T.F.M.et al (2005) изучив результаты МРТ исследования 6 пациентов с Mobius syndrome пришли к выводу, что он является частью более сложного порока, включающего аномалию структур задней черепной ямки (DWC), выявляемую у 50% пациентов.
Известна роль мозжечка в контроле и интеграции моторной деятельности. Однако, его многочисленные связи с ассоциативными и паралимбическими областями коры мозга, лежат в основе влияния мозжечка на познавательную деятельность, регуляцию эмоций, тонкой моторики, экспрессивной речи, устной памяти, способности к планированию (Schmahmann J., 1991; Калашникова Л.А., 2001; Philip N et al, 2003).
Первую попытку исследования зависимости между степенью дисплазии мозжечка, его функцией и интеллектом при пороке Денди–Уокера предприняли Gerszten PC et al в 1995 году. Они ретроспективно изучили 20 пациентов с DWC. Морфометрическое исследование мозжечка включало измерение его размеров и размеров задней черепной ямки по результатам компьютерной томографии, а так же, отношение мозжечкового размера к размеру задней черепной ямки. Нормальная функция мозжечка была диагностирована у 50 % больных и нормальный интеллект - у 45 % пациентов. Корреляция между размером мозжечка, его функцией и интеллектуальным развитием не была установлена.
Klein O, Pierre-Kahn A et al (2003) проведено исследование, целью которого явилось выявление взаимосвязи между вариантами структурных нарушений при синдроме Денди–Уокера и прогнозом неврологического и психического развития. В исследование были включены 26 пациентов с DWM имеющие следующие нарушения: 1) большую кисту задней черепной ямки, широко сообщающуюся с IV желудочком; 2) гипопластичный, смещенный вверх червь мозжечка; 3) смещенный вверх намет мозжечка; 4) увеличенную заднечерепную ямку; 5) нормальные полушария мозжечка; 6) нормальный ствол мозга. Изучалась зависимость интеллектуального развития от особенностей строения червя мозжечка, размеров желудочков мозга, анатомии мозга и аномалий других органов. Исследование сагиттальных Т2-взвешенных изображений позволило выделить две группы пациентов. В первой (n=21) – червь мозжечка имел две борозды, три дольки и нормально расположенные ядра шатра. Ни один из пациентов этой группы не имел других пороков развития мозга и все пациенты, кроме двух имели нормальное психическое развития (у одного из двух умственно отсталых детей диагностирован синдром фрагильной X хромосомы, у другого тяжелая перивентрикулярная лейкомаляция вследствие преждевременного рождения). Во второй группе (n=5), червь мозжечка был диспластичен, имел не более одной борозды, разделение его на доли было нарушено. Нарушение строения червя мозжечка в этой группе постоянно сочеталось с другими аномалиями мозга, наиболее часто с агенезией мозолистого тела. Все пациенты второй группы имели различную степень умственной отсталости. Таким образом, анатомия червя мозжечка статистически коррелировала с неврологическим и интеллектуальным развитием пациентов.
Работы Boddaert N et al (2003) подтверждают корреляцию анатомического строения червя мозжечка и интеллектуального развития.
Нормальное интеллектуальное развитие обнаружено у 50-75% пациентов с аномалией Денди–Уокера (Genevieve Lefort et al, 2001; Klein O et al, 2003), нарушения психического развития связывают с сочетанием порока с другими нарушениями развития ЦНС, встречающимися у 68% пациентов. Они включают гипоплазию мозолистого тела, встречающуюся у 17–25% пациентов (Sawaya R et al, 1981; Barkovich A. James et al, 2001), нарушения нейрональной миграции в коре больших полушарий и коре мозжечка (дисплазию поясной извилины – 25%; полимикрогирию – 5–10%; шизэнцефалию, мозжечковую дистопию), голопрозэнцефалию у 25% (Yamashita N et al, 1992), пороки нижней оливы, энцефалоцеле, гипоплазию стволовых структур, сирингомиелию, менингоцеле в затылочной, пояснично-крестцовой области, спинномозговую липому (Tan EC et al, 1995; Saatci I et al, 1998; Shuto T et al, 1999; Ben Hamouda H et al, 2001; Joy HM et al, 2001; D.P. Muzumdar et al, 2004; Smith AS, Levine D, 2004).
Soto-Ares G (2000) изучил результаты МРТ- исследования 17 пациентов с мозжечковой корковой дисплазией. У двух из них порок был изолированным. У 11 пациентов (65%) мозжечковая корковая дисплазия сочеталась с нарушением развития червя мозжечка, широко распространенный порок развития задней черепной ямки (порок Денди–Уокера) наблюдался у 8 пациентов (47%). Клинические нарушения были представлены выраженной задержкой психического и двигательного развития, мышечной гипотонией, лицевым дизморфизмом, патологией глаз или глазниц. У пациентов часто отмечалось косоглазие, нистагм, скелетные деформации, деформации пальцев: брахидактилия и клинодактилия.
Sandeep Patel and A.James Barkovich (2002) изучили 70 МРТ изображений аномалий мозжечка. Пациенты с пороком Денди–Уокера (n=19) имели и гипоплазию, и дисплазию мозжечка. Все пациенты с диффузной мозжечковой дисплазией имели дисплазию полушарий головного мозга. При фокальной мозжечковой дисплазии вследствие генетических синдромов (Joubert и rhombencephalosynapsis), дисплазия полушарий большого мозга могла определяться или отсутствовать. Пациенты с несиндромной фокальной мозжечковой дисплазией (изолированная фокальная мозжечковая корковая дисплазия, мозжечковая дистопия с мозжечковой корковой дисплазией, идиопатическая рассеянная мозжечковая дисплазия, изолированная мозжечковая гипоплазия и понтоцеребеллярная гипоплазия) имели нормальные большие полушария мозга. Не были обнаружены различия возможных вариантов нарушений строения ЦНС у пациентов с кистами четвертого желудочка (DWM) и изолированной мозжечковой гипоплазией. В связи с этим Sandeep Patel and A.James Barkovich указывают, что порок Денди–Уокера является гетерогенным.
У 20-33% пациентов встречается сочетание аномалии Денди–Уокера с пороками развития других органов и систем: аномалиями черепа и позвоночника типа Клиппеля–Фейля, расщелиной губы и твердого неба, пороками сердца и сосудов (Mohammadi M et al, 2004), мочевыводящих путей (Alanay Y et al, 2005). Pillay K et al (2003) описывают DWM у плода с фацио-аурикуло-вертебральной последовательностью, сочетанием синдромов Rokitansky (атрезия влагалища, двурогая матка, аномалии мочевыводящей системы) и DiGeorge (аплазия тимуса, гипопаратиреоидизм и порок сердца).
Нарушения в строении и функции других органов и систем в сочетании с классической триадой порока Денди–Уокера называют синдромом Денди–-Уокера (DWMS), этиология синдрома всегда требует уточнения и исключения наследственного генеза.
Большинство пороков развития Денди–Уокера является спорадическими. База данных Possum сообщает о 6 аутосомно-доминантных и 37 аутосомно-рецессивных синдромах, которые могут быть связаны с комплексом Денди–Уокера, особенностью этих синдромов является сочетание с множественными аномалиями внутренних органов (Genevieve Lefort et al, 2001). Wakeling EL et al (2002) сообщают о случае X-сцепленного наследования варианта Денди-Уокера.
Bordarier и Aicardi выделяют синдромы, при которых гипоплазия червя мозжечка является одним из определяющих признаков, такие как Joubert's syndrome (Maria BL et al, 2001), Walker Warburg syndrome, muscle-eye-brain syndrome (MEB), Goldston syndrome, PHACE syndrome и гипоплазия червя мозжечка с колобомой радужки и фиброзом печени. В ряде других случаев гипоплазия червя мозжечка может встречаться, однако, не является закономерной: Fukuyama congenital muscular dystrophy, Meckel–Gruber (Al-Gazali et al, 1996; Balci S et al, 2004; Nizard J et al, 2005), orofacial digital syndrome type II (Toriello HV et al, 2002), Coffin Siris syndrome (Fleck B. J. et al, 2001; Imai T et al, 2001), Smith Lemli Opitz syndrome, Ellis van Creveld syndrome, Ruvalcaba Myhere Smith syndrome, Kallmann syndrome (Ueno H et al, 2004), cri-du-chat syndrome (De Michele G et fl, 1993).
Генерализованная церебеллярная гипоплазия с нормальным IV желудочком
В классификации аномалий мозжечка, предложенной Patel S и Barkovich AJ. (2002) как подкласс выделена генерализованная гипоплазия мозжечка без расширения задней черепной ямки и кисты IV желудочка. К этой группе относятся все случаи сочетанной гипоплазии червя и полушарий мозжечка разной степени выраженности. В группе выделены два подкласса в зависимости от наличия или отсутствия гипоплазии структур ствола и продолговатого мозга (Рис.5, 6). Barkovich AJ. обращает внимание достаточно хорошее двигательное и интеллектуальное развитие пациентов. В его исследовании три человека, имевших генерализованную гипоплазию мозжечка, ассоциированную с выраженной гипоплазией ствола и продолговатого мозга были направлены для обследования в связи с жалобами на легкую атаксию в 2-х случаях, и головную боль в одном случае.
Подробный обзор синдромов, включающих гипоплазию мозжечка, проведен Boltshauser E. (2008).
Рис.5. Генерализованная гипоплазия мозжечка и ствола мозга.
Рис.6. Генерализованная гипоплазия мозжечка.
В литературе отсутствуют наблюдения полной церебеллярной агенезии, вероятно, она не совместима с жизнью. Однако, имеется достаточное число описаний выраженной церебеллярной гипоплазии. Данный порок выявлен Gardner et al. (2001) у пяти детей, во всех случаях определялась редуцированная и деформированная ткань мозжечка, обычно, сохранными оставались нижние ножки, передние дольки червя и клочок мозжечка (Zafeiriou et al., 2004). Несколько пациентов с данной патологией были описаны в исследовании Richter et al. (2005), все имели психомоторные нарушения различной степени тяжести, но смогли освоить ходьбу без опоры. Все случаи изолированной гипоплазии мозжечка являются спорадическими. Патогенез генерализованной церебеллярной гипоплазии до конца не понятен и, вероятно, мультифакториален. Гены, ответственные за возникновение порока в настоящее время не картированы. Мутация гена Engrail ed-1 у мышей ведёт к аплазии мозжечка и структур среднего мозга, но сопровождается 100% летальностью (Bilovocky et al., 2003).
X-сцепленная церебеллярная гипоплазия. Фенотипические проявления синдрома включают макроцефалию, диспластичное лицо, высокий рост, гипогенитализм. Клинические признаки заболевания представлены диффузной мышечной гипотонией, задержкой развития психомоторных функций и страбизмом, с возрастом присоединяется миоклоническая эпилепсия, появляются признаки умственной отсталости, степень выраженности атактического синдрома вариабельна (Renier et al., 1983; Philip et al., 2003). Фенотипические особенности женщин носительниц мутантного гена вариабельны и могут включать как полное отсутствие признаков патологии, так и явления умеренного лицевого дизморфизма, нарушение когнитивных функций.
У большинства пациентов нейровизуализация выявляет супратенториальные структурные изменения в виде расширения экстрацеребральных пространств, вентрикуломегалии, атрофии nuclei caudate. Характерные субтенториальные структурные нарушения включают различную степень гипоплазии червя и полушарий мозжечка с расширением большой затылочной цистерны.
В нескольких семьях выявлены мутации oligophreni n-1гена (OPHN 1) на хромосоме Xq12 (Bergmann et al., 2003; Philip et al., 2003; des Portes et al., 2004). Следует отметить, что мутация гена OPHN1 часто определяется при несиндромальной форме умственной отсталости и ген OPHN1, очевидно, играет значимую роль в процессах формирования мозжечка. В тоже время, в литературе имеется описание двух семей с X-сцепленной церебеллярной гипоплазией не связанной с мутацией гена OPHN1, что говорит о гетерогенности патологии (Philip et al., 2003).
PHACE syndrome. Церебеллярная гипоплазия с гемангиомой лица впервые описана Pascual-Castroviejo I (1978). В 1981г. Sawaya и McLaurin, анализируя собственные наблюдения и данные литературы, обратили внимание на ассоциацию между Dandy-Walker malformation и гемангиомами лица. Позже Frieden IJ с соавторами (1996) была предложена аббревиатура PHACE для обозначения синдрома включающего мальформации задней черепной ямки, гемангиомы, аномалию артерий, коарктацию аорты, врождённый порок сердца и патологию глаз (posterior fossa malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects and eye abnormalities). Многие исследования подтверждают гетерогенность синдрома, возможно отсутствие в его структуре одного или более компонентов (Metry et al., 2001). Определяющими признаками заболевания являются гемангиомы лица и аномалии мозжечка, которые могут быть представлены DWM или церебеллярной гипоплазией (часто асимметричной), церебеллярной кортикальной дисплазией (Rossi et al., 2001; Bhattacharya et al., 2004; Boltshauser, 2004).
Рис.7. Гипоплазия мозжечка у ребёнка с PHACE syndrome.
Сагиттальное и фронтальное T1-weighted изображение асимметричной гипоплазии червя и полушарий мозжечка при нормальных размерах задней черепной ямки и ствола мозга у ребёнка с PHACE syndrome (собственные данные Клеймёнова И.С., 2010).
Poetke M et al. (2002) изучив сообщения о 59 пациентах с PHACE синдромом и десять собственных случаев, приводят следующие результаты: наиболее частой аномалией ЦНС был DWS диагностированный у 81% пациентов, пороки развития артерий были найдены в 22% случаев, только 19% пациентов имелись пороки развития крупных сосудов без синдрома Денди-Уокера, треть больных имела аномалии глаз, сердца, аорты. Гемангиомы языка были выявлены у 7% пациентов. У 12% пациентов с PHACE синдромом диагностированы внутричерепные гемангиомы. Развитие когнитивных функций не прослежено в большинстве сообщений, однако, Boltshauser E (2008) описывает пациента с данным синдромом не имевшего когнитивных расстройств в школьном возрасте.
Ritscher–Schinzel syndrome. Синдром был описан у двух сестёр Ritscher и соавторами в 1987 году. Одна из девочек погибла, вероятно, вследствие дисфункции вентрикулярного шунта, у второй с возрастом выявились когнитивные нарушения и иммунодефицит (Zankl et al., 2003). Нейровизуализация выявила у детей Dandy-Walker malformation ассоциированную с пороком сердца. Verloes и соавторы (1989) применили термин "3C syndrome" обозначивший кранио-церебелло-кардиальную дисплазию (cranio-cerebello-cardiac dysplasia). Пациенты с Ritscher–Schinzel синдромом имели гипоплазию мозжечка, гипертелоризм, антимонголоидный разрез глаз, низко расположенные ушные раковины, постнатальную задержку роста, пороки сердца, нарушение развития психомоторных функций. В структуре заболевания описаны одностороння потеря слуха и дефицит гормона роста (Wheeler et al., 1999). Donahue и Ryan (1995) приводят данные о случае подтверждённой делеции в хромосоме 8q21-22 у пациента с синдромом Ritscher–Schinzel.
Hoyeraal–Hreidarsson syndrome. В 1988 году Hreidarsson с соавторами описали мальчика умершего в возрасте 23 месяцев. Он страдал врождённой тромбоцитопенией, имел микроцефалию, нейровизуализация выявила признаки гипоплазии мозжечка и перивентрикулярные кальцификаты. Погиб ребёнок вследствие прогрессирующего инфекционного процесса на фоне панцитопении. Hreidarsson отметил, что описание подобного наблюдения было сделано Hoyeraal с соавторами в 1970 году. Hoyeraal–Hreidarsson syndrome – мультисистемное заболевание характеризующееся врождённой задержкой роста, вторичной микроцефалией, нарушением развития психомоторных функций, атаксией, иммунодефицитом, прогрессирующими изменениями в костном мозге с развитием панцитопении, описаны генерализованная лимфоаденопатия и гепатоспленомегалия. Синдром наследуется по X-сцепленному типу, связан с мутацией гена DKC1 на хромосоме Xq28 (Sznajer et al., 2003). Типичным признаком заболевания является церебеллярная гипоплазия, гистологическое исследование выявляет нарушение организации коры мозжечка с гипоплазией гранулярного слоя (Berthet et al., 1994).
Врождённая непрогрессирующая атаксия. К данной группе относятся пациенты, не имеющие признаков инфекционного поражения ЦНС, перенесённого перинатального инсульта, мальформации мозжечка, в том числе идентифицированных наследственных синдромов (Steinlin et al., 1998b). Клинические признаки заболевания включают врождённую непрогрессирующую атаксию, маскирующуюся мышечной гипотонией в раннем возрасте, а также задержку развития психомоторных функций. Типичные симптомы поражения мозжечка, включающие туловищную атаксию, интенционный тремор, дизартрию, появляются с возрастом по мере созревания ЦНС. У всех пациентов наблюдается нарушение когнитивных функций различной степени тяжести (Schmahmann and Sherman, 1998; Steinlin et al., 1998b, 1999; Wassmer et al. (2003), отмечается высокая частота судорожных приступов (Parmeggiani et al., 2003), коме того имеются сведения об аутистическом поведении. Нейровизуализация не выявляет выраженного уменьшения объёма мозжечка, но часто определяется расширение борозд между извилинами, что заставляет проводить дифференциальный диагноз с атрофией мозжечка (Margari et al., 2004). Многие авторы приводят данные о повторных случаях заболевания в одной семье (Steinl in et al., 1998b; Margari et al., 2004), однако, гены вызывающие врождённую непрогрессирующую атаксию не картированы. Предполагается возможность наследования сцепленного с 9q хромосомой (MIM 213200) (Megarbane. et al., 1999; Delague et al., 2001), с 19 хромосомой (Nystuen et al., 1996). Идентифицирован ген (Caytaxin, MIM 608 179), мутация которого вызывает атаксию и судороги у мышей, он вызывает нарушение функции кальциевых каналов, однако, у человека данная мутация не известна (Brill et al., 2004). Описано возникновение аутосомно-рецессивной церебеллярной гипоплазии в результате делеции рецепторов липопротеидов низкой плотности (VLDLR) (Boycott et al., 2005).
Гипоплазия мозжечка связанная с болезнями нарушения обмена
Гипоплазия мозжечка как следствие болезней обмена встречается редко, в то время как атрофия мозжечка с поражением белого вещества и церебеллярных ядер характерный признак многих метаболических расстройств (Steinlin et al., 1998).
Врождённое нарушение гликозилирования представляет собой группу аутосомно-рецессивных заболеваний связанную с патологией генов ответственных за процесс N-гликозилирования. Наиболее часто встречается тип Ia для которого характерна глобальная церебеллярная гипоплазия, сопровождающаяся атрофией вещества мозжечка. Встречающаяся, как правило, гипоплазия моста и продолговатого мозга создают трудности при проведении дифференциального диагноза между нарушением гликозилирования и pontocerebellar hypoplasia type 2. Характерным признаком нарушения гликозилирования IId типа является гипоплазия мозжечка с формированием DWM (Peters et al., 2002).
Smith–Lemli–Opitz syndrome относится к болезням нарушения обмена ассоциированным со структурными аномалиями мозга и связано с нарушением синтеза холестерола (Kelley and Hennekam, 2000). Выделено два типа заболевания, один из которых (тип II) заканчивается летально в неонатальном периоде. Фенотипические признаки синдрома включают микроцефалию, птоз, широкий кончик носа, микрогнатию, описана синдактилия, костные деформации конечностей, расщелина нёба, аномалии лёгких, поджелудочной железы, пилоростеноз. Нейровизуализация выявляет гипоплазию мозжечка. Irons et al. (1993) и Tint et al. (1994) отметили выраженное снижение концентрации холестирола в плазме крови и повышение уровня 7-дегидрохолестерола. Tint et al. (1995) исследовав 24 пациента с первым типом синдрома, подтвердили прямую корреляцию между тяжестью заболевания и концентрацией холестерола в плазме.
Рис. 8. Синдром Smith–Lemli–Opitz.
Фенотип ребёнка с Smith–Lemli–Opitz syndrome. В неврологическом статусе определялась микроцефалия, центральный тетрапарез, резистентная симптоматическая эпилепсия, грубое нарушение психомоторных функций. МРТ исследование головного мозга выявило лиссэнцефалию, ассоциированную с гипоплазией червя и полушарий мозжечка. (собственные данные, Клеймёнова И.С., 2010).
Дефицит адренолозуциназы относится к редкой патологии ассоциированной с умеренной церебеллярной гипоплазией (Kohler et al., 1999). Клинические признаки заболевания включают выраженное нарушение психомоторного развития, эпилепсию и аутистикоподобное поведение (Boltshauser E., 2008).
Гипоплазия мозжечка при митохондриальных болезнях встречается редко, однако, описаны случаи церебеллярной гипоплазии при болезни дыхательных цепей и дефиците пируватдегидрогеназы (Lincke et al., 1996).
Церебеллярная гипоплазия не является характерным признаком нейрофиброматоза, однако, в литературе встречаются описания гипоплазии мозжечка при нейрофиброматозе 1 типа (Boltshauser E., 2008).
В соответствии с классификацией Barkovich AJ. (2002) К группе генерализованных церебеллярных гипоплазий с нормальным IV желудочком следует относить дегенеративные заболевания с прогрессирующей атрофией мозжечка.
В частности к данному классу относится аутосомно рецессивное дегенеративное заболевание с пренатальным началом Pontocerebellar Hypoplasia Barth. Заболевание впервые описано Norman и Urich in 1958, относится к дегенеративным и большинство пациентов погибают в детстве. Следует отметить, что важным признаком патологии является прогрессирующая гипоплазия полушарий мозжечка при нормальном черве. Вентральная часть моста значительно уменьшена в размерах, так же структурные изменения выявляются в нижних оливах, ядрах таламуса, возможны негрубые атрофические изменения коры больших полушарий мозга. Данную патологию Barth наблюдал в Датской популяции у сибсов, имевших микроцефалию, пирамидные и экстрапирамидные расстройства, посмертное патологоанатомическое исследование которых выявило признаки значительных нейрональных потерь в структурах моста и мозжечка пациентов (Barth, 1993; Barth et al., 1995). В 1995 году Barth et al. выделил два типа патологии:
Тип I (PCHI) сопровождался дегенеративными изменениями передних рогов спинного мозга. Клинические проявления заболевания включали микроцефалию, множественные врождённые контрактуры суставов, грубое нарушение психомоторного развития. Дети погибали на первом году жизни от прогрессии дегенеративного процесса в ЦНС, сопутствующих бульбарных, дыхательных расстройств и инфекционных осложнений.
Тип II (PCHII) объединил расстройства, вызванные дегенеративными изменениями с преимущественной локализацией в области ствола и полушарий мозжечка. Клинические проявления включают микроцефалию, бульбарные расстройства, проявляющиеся трудностями при вскармливании, судорожные приступы, дистонические реакции. Пациенты доживают до пубертатного возраста.
Нейровизуализация может выявить структурные изменения подобные PCH при ряде других заболеваний: врождённые нарушения гликозилирования, врождённая мышечная дистрофия (Walker–Warburg syndrome, MEB), митохондриальных болезнях, субтотальной церебеллярной агенезии, повреждениях мозжечка у глубоко недоношенных детей (Johnsen et al., 2005). Гены ответственные за развитие заболевания в настоящее время не установлены.
Генерализованная гипоплазия мозжечка характерна для спинальной мышечной атрофии с понтоцеребеллярной гипоплазией (Spinal muscular atrophy – pontocerebellar hypoplasia). Данное заболевание трудно для дифференциальной диагностики с Werdnig-Hoffman disease: у новорождённых младенцев определяется глубокая мышечная гипотония, фасцикуляции в языке. Однако, в отличие от спинальной мышечной атрофии Werdnig-Hoffman наблюдаются нистагмоидные движения глазных яблок связанные с корковой слепотой, возможно присоединение моторной и сенсорной невропатии. Посмертное вскрытие помимо дегенеративных изменений в спинном мозге выявляет атрофию мозжечка с вовлечением в патологический процесс моста, продолговатого мозга. Тип наследования рецессивный. Исследование пораженных семей позволяет выявить случаи мышечной гипотонии и контрактур суставов у родственников.
Фокальная гипоплазия мозжечка
Изолированная гипоплазия одной гемисферы. Первое описание унилатеральной церебеллярной гипоплазии датируется 1915 годом, когда Strong описал данную мальформацию у 3-х летней девочки умершей от кори и имевшей грубое нарушение психомоторного развития с раннего возраста. У девочки были выявлены ассоциированные аномалии, включавшие гипоплазию ипсилатеральных верхних и средних ножек мозжечка, верхних холмиков четверохолмия, асимметричный мост с отсутствием контралатеральных олив. Нарушения психомоторного развития при унилатеральной церебеллярной гипоплазии описаны (Boltshauser et al., 1996, 2008).
Три пациента (два ребёнка и 1 взрослый) с изолированной унилатеральной гипоплазией полушария мозжечка описаны в исследовании Patel S, Barkovich AJ (2003). Причиной направления детей на обследование было нарушение развития психоречевых функций, взрослый пациент страдал эпизодическими головными болями. Неврологическое обследование выявило у всех пациентов симптомы «мягкой» атаксии, другие очаговые симптомы выявлены не были. Нарушений строения червя мозжечка, ствола, продолговатого мозга, контралатеральной гемисферы мозжечка, а также полушарий большого мозга у пациентов этой группы выявлено не было.
Изолированная унилатеральная церебеллярная гипоплазия, выявленная при УЗ исследовании головного мозга и подтвержденная при проведении МРТ плоду на 20–24 недели гестации описана Robins et al. (1998). Отсутствие признаков перенесённого кровоизлияния свидетельствовало о врождённом характере мальформации. Однако, в современной литературе продолжает обсуждается вопрос, является ли данный порок мальформацией или дизрубцией. Возможно, генетический механизм формирования данного порока тот же, что и при формировании тотальной агенезии мозжечка?
Рис. 9. Унилатеральная церебеллярная гипоплазия у 29-летней женщины.
Аксиальное T1-weighted изображение, демонстрирует гипоплазию правой гемисферы мозжечка при нормальных размерах червя и правой гемисферы (Patel S, Barkovich AJ, 2003).
Изолированная гипоплазия червя мозжечка. Изолированная гипоплазия нижних отделов червя мозжечка, не всегда рассматривается как аномалия (Patel S, Barkovich AJ, 2003), поскольку часто данное нарушение является случайной находкой при проведении нейровизуализации и пациенты, с гипоплазией нижних отделов червя не имеют двигательных и психических расстройств.
Однако данное нарушение выявляется при некоторых синдромах, сопровождающихся нарушением двигательного и психического развития. В этих случаях гипоплазия червя может сочетаться с супратенториальными аномалиями.
Например, гипоплазия червя мозжечка ассоциированная с дисгенезией мозолистого тела описана у ребенка с синдромом «кошачьего крика», Исследование кариотипа выявило делецию короткого плеча 5 хромосомы. Неврологический осмотр выявил нарушение развития психоречевых функций, поведения, признаки мозжечково-спинального повреждения (De Michele G et al, 1993). У пациентов с мерозин дефицитной мышечной дистрофией наблюдалось нарушение миелинизации проводников со снижением плотности белого вещества больших полушарий и мозжечка, гипоплазией нижних отделов червя мозжечка. Нарушений строения коры мозжечка выявлено не было (Patel S, Barkovich AJ, 2003).
II. Дисплазия мозжечка
Генерализованная дисплазия
Врожденная мышечная дистрофия. Среди врождённых мышечных дистрофий выделяют две подгруппы:
В связи с общностью механизмов возникновения для всех случаев характерна диффузная мышечная гипотония со снижением рефлексов, выявляются аномалии ЦНС, различающиеся по тяжести проявлений. У пациентов с Fukuyama congenital muscular dystrophy и Walker-Warburg syndrome описана дисплазия коры полушарий мозжечка по типу «булыжной мостовой», в ряде случаев диагностирована полимикрогирия (van der Knaap MS, Smit LM, Barth PG, et al, 1997; Barkovich AJ., 1998; Farina L, Morandi L, Milanesi I, et al.,1998; Lamer S, Carlier RY, Pinard JM, et al, 1998).
Подгруппа мышечных дистрофий, ассоциированных с мальформациями мозга включает:
Пациенты с WWS, MEB, Fukuyama CMD с рождения имеют глубокую мышечную гипотонию, грубое нарушение психомоторных функций, высокий уровень креатинкиназы, структурные аномалии головного мозга и глаз. Клинические признаки CMD 1B,1C, 1D и limb girdle muscular dystrophy 2I менее серьёзны.
Рис.10. Пациент страдающий Walker-Warburg syndrome и результаты МРТ головного мозга .
Описанный в 1971 году Walker-Warburg syndrome (WWS) является аутосомно-рецессивным расстройством и включает сочетание глазных аномалий (дисплазию сетчатки, отслойку сетчатки, микрофтальмию, вторичные катаракты) и пороки развития мозга (лиссэнцефалию II типа, гипо- или аплазию мозолистого тела и прозрачной перегородки, окклюзионную гидроцефалию, аплазию червя мозжечка и гипоплазией/дисплазией его полушарий и ствола мозга) и врождённую мышечную дистрофию (CMD). Walker-Warburg syndrome ассоциирован с мутациями в генах POMT1, POMT2, FKRP, Fukutin. В 7–20% случаев определяется мутация гена POMT1, расположенного в хромосоме 9q34. Клиническая картина заболевания представлена окклюзионной гидроцефалией, диффузной мышечной гипотонией со снижением сухожильных рефлексов и повышением уровня креатинкиназы, умственной отсталостью, судорогами.
Болезнь мышца-глаз-мозг (muscle-eye-brain disease – MEB) проявляется в течение первых шести месяцев жизни в виде тяжелой прогрессирующей мышечной гипотонии, повышения уровня креатинкиназы в сыворотке крови, развития гидроцефального синдрома, задержки психо-моторного развития, патологии глаз (плавающие движения глазных яблок, крупноразмашистый нистагм, дефекты передней камеры с глаукомой, близорукость, дистрофия сетчатки, вторичная катаракта). Результаты ЭНМГ исследования и биопсии мышечной ткани характерны для прогрессирующей мышечной дистрофии. МРТ головного мозга выявляет лиссэнцефалию II типа, агенезию мозолистого тела, гипоплазию ствола мозга и дисплазию мозжечка.
В связи со сходством фенотипических проявлениях WWS и MEB клиническая дифференциальная диагностика невозможна. Однако тяжесть неврологических расстройств меньше при WWS. Прогноз при WWS более благоприятный, но глазные аномалии (дистрофия сетчатки, катаракта) являются не врожденными, а прогрессирующими. Cormand B. et al. (2001) проведены исследования 29 пациентов с фенотипом MEB/WWS, свидетельствующие о генетической гетерогенности заболеваний. При MEB-фенотипе доказана связь с 1p32-p34, у пациентов с фенотипом WWS такой связи не выявлено. Таким образом, в настоящее время дифференциальный диагноз между WWS и MEB базируется на генетических исследованиях: в большинстве случаев muscle-eye-brain disease является следствием мутации в гене POMGnT1, при Walker-Warburg syndrome чаще определяются мутации в гене POMT1 (Uluc Y et all, 2007).
Фенотипическое сходство с MEB и WWS имеется и при врожденной мышечной дистрофии Фукуямы (Fukuyama CMD). Это аутосомно-рецессивное заболевание, вызванное мутацией в гене fukutin расположенном на 9q хромосоме, типично для Японии и впервые описано в 1960г. Fukuyama. Белок, кодируемый данным геном, представлен в аппарате Гольджи клеток скелетных мышц, сердца, головного мозга. Электронная микроскопия выявляет разрушение базальной мембраны мышечных клеток при данной патологии.
Рис.11. Пациент с muscle-eye-brain disease, результаты МРТ головного мозга.
Типичными клиническими признаками являются низкая двигательная активность плода, слабое сосание, отсутствие попыток удержать голову в неонатальном периоде. В связи с врождённой мышечной слабостью пациенты изредка могут самостоятельно стоять и непродолжительное время самостоятельно ходить.
Рис.12. Пациент с Fukuyama CMD.
Характерно прогрессирование мышечной слабости и формирование контрактур с возрастом. У большинства пациентов после 10 лет жизни развивается дилатационная кардиомиопатия. Биопсия мышц позволяет выявить их дистрофические изменения. Зрительные расстройства включают миопию, гипоплазию зрительных нервов, микрофтальмию, катаракту. Отмечается умственная отсталость тяжелой степени, 40% пациентов имеют судорожные приступы. Смерть, как правило, наступает в возрасте 10–20 лет. Однако, имеются сообщения о субклиническом проявлении нарушений развития психических функций даже при наличии структурной патологии со стороны ЦНС.
Результаты МРТ головного мозга выявляют дефекты нейрональной миграции с формированием лиссэнцефалии или полимикрогирии, дистопии пирамидных трактов в стволе головного мозга, вентрикуломегалию, атрофию коры головного мозга, снижение плотности белого вещества, синдром Денди-Уокера, кисты и дисплазию мозжечка.
Врождённая цитомегаловирусная инфекция. Проявления аномалий развития ЦНС вследствие врождённой цитомегаловирусной инфекции сходны в полушариях большого мозга и мозжечка. Описаны лиссэнцефалия с истончением коры головного мозга, маленьким «сморщенным» мозжечком с истончённой корой и мелкими бороздами. Диагностировалась атрофия гемисфер мозжечка, червь не визуализировался, ствол мозга был гипопластичен. В ряде случаев у пациентов с диффузной полимикрогирией мозжечок был нормальных размеров, с негрубыми признаками дисплазии (нарушение ориентации извилин).
Рис.13. Дисплазия мозжечка у ребёнка с врождённой цитомегаловирусной инфекцией (Patel S, Barkovich AJ, 2003).
Коронарное T1-weighted изображение, демонстрирует выраженное уменьшение размеров мозжечка с признаками его дисплазии. Гиперинтенсивные сигналы в правой его гемисфере являются результатом кальцификации. Кора больших полушарий изменена по типу лиссэнцефалии.
В одном случае (Patel S, Barkovich AJ, 2003) выявлена билатеральная гетеротопия в мозговом теле мозжечка (corpus medullare), ассоциированная с нарушением ламинации в обеих гемисферах. Кроме того, у пациента имелись супратенториальные аномалии развития мозга в виде снижения плотности белого вещества мозга, субкортикальной гетеротопии ассоциированной с полимикрогирией. Структура и размеры мозолистого тела не были изменены.
Генерализованная дисплазия мозжечка ассоциированная с лиссэнцефалией. Лиссэнцефалия – порок, формирующийся в период 2–3 месяца внутриутробного развития и характеризующийся значительным уменьшением или отсутствием извилин. Степень нарушения может варьировать от агирии (отсутствие извилин и борозд, трёхслойный, дезорганизованный неокортекс с множественными перивентрикулярными нейронными гетеротопиями) до пахигирии (небольшое число плоских расширенных извилин, четырёхслойное строение неокортекса). Лиссэнцефалия является результатом нарушения поздней волны миграции нейронов из герминативного матрикса в область коры головного мозга. В настоящее время группа лиссэнцефалий поделена на синдромы, каждый из которых характеризуется мутацией определённого гена (LISI, DCX, RELN).
1. Классическая лиссэнцефалия (1-й тип):
а) связанная с 17-й хромосомой:
б) X – связанная;
в) другой локализации.
2. Лиссэнцефалия по типу «булыжной мостовой» (2-й тип):
а) врождённая мышечная дистрофия Фукуямы;
б) синдром Уокера-Варбурга;
в) болезнь «мышца-глаз-мозг».
3. Неклассифицированные лиссэнцефалии.
Классическая лиссэнцефалия 1-го типа характеризуется гладкой поверхностью коры головного мозга, при её четырёхслойном строении. Нейровизуализация выявляет агирию лобных и пахигирию теменно-затылочных отделов мозга, вертикально расположенные расширенные сильвиевы борозды. Объём белого вещества мозга уменьшен, при нейровизуализации выявляется прямолинейный характер границы между серым и белым веществом, ленточная гетеротопия нейронов. Может определяться гипоплазия мозолистого тела, ствола мозга.
Лиссэнцефалия 1-го типа встречается при изолированном типе лиссэнцефалии, в 20–30% связана с делецией 17p и в 40% – с мутацией гена LISI, а также при аутосомно-доминантном синдроме Миллера-Дикера. Для последнего характерен выраженный черепно-лицевой дизморфизм (микроцефалия, запавшая переносица, гипоплазия срединной части лица), врождённый порок сердца, постаксиальная полидактилия. Нейровизуализация выявляет тяжёлую лиссэнцефалию с очагом кальциноза по средней линии. В 50–70% случаев синдрома Миллера-Дикера найдена делеция 17p13.3.
Лиссэнцефалия по типу «булыжной мостовой» (2-й тип) впервые описана Уокером в 1942 году. Проявляется гладкой, утолщённой корой головного мозга с нарушением её цитоархитектоники, аномалией кровеносных сосудов, дезорганизацией фиброглиальных волокон, выраженной гипомиелинизацией белого вещества мозга и нечеткостью границ между серым и белым веществом. Порок связан с нарушением разделения мезенхимы и клеток нервной системы. Главной причиной лиссэнцефалии II типа является повреждение экстрацеллюлярного матрикса. В результате нарушения структуры поверхностной базилярной мембраны происходит эктопия в мягкие мозговые оболочки нейронов и глиальных клеток. Выявлено изменение I, III, VI подтипов коллагена у пациентов с данным пороком. Аномалия ассоциирована с гидроцефалией, агенезией или гипоплазией мозолистого тела, гипоплазией червя мозжечка. Этот тип лиссэнцефалии выявлен при мышечной дистрофии Фукуямы, синдроме Уокера–Варбурга, болезни «мышца-глаз-мозг».
Рис.14. Лиссэнцефалия и церебеллярная гипоплазия у новорождённого с мутацией RELN (Patel S, Barkovich AJ, 2003).
А – сагиттальное T1-weighted изображение мальформации мозга по типу лиссэнцефалии, гипоплазии/дисплазии мозжечка и ствола мозга.
В - Аксиальное T1-weighted изображение демонстрирует гипоплазию/дисплазию гемисфер мозжечка, гипопластичный ствол с центральной расщелиной.
Лиссэнцефалия с мутацией RELN. Hong SE et al (2000) описана аутосомно-рецессивная форма лиссэнцефалии ассоциированная с тяжёлыми аномалиями мозжечка, гиппокампа и ствола мозга. Мутантный ген, кодирующий релин (RELN), картирован на 7q22 хромосоме. Данная мутация ведёт к повреждению RELN cDNA, результатом чего является резкое снижение синтеза релин-протеина. Reelin представляет собой экстрацеллюлярный матрикс-ассоциированный протеин, играющий важную роль в обеспечении нейрональной миграции в период развития коры головного мозга (Chang BS et al, 2007). Этот белок имеет молекулярную массу около 388 kD и участвует в связывании нейронов коры головного мозга с рецепторами липопротеинов низкой плотности (VLDLR), аполипопротеином Е рецептора 2 (ApoER2; refs 9–11), alpha3beta1 интегрином. Полученная в эксперименте подобная мутация у мышей ведёт к церебеллярной гипоплазии, аномалиям нейрональной кортикальной миграции и нарушению формирования синаптических контактов.
Спектр кортикальных мальформаций таких как лиссэнцефалия ассоциированная с аномалиями мозжечка, называется лиссэнцефалия с церебеллярной гипоплазией (lissencephaly with cerebellar hypoplasia – LCH). Ross ME et al (2001) обследовали 34 ребёнка, имевших данные расстройства, что позволило выделить 6 подтипов LCH. Два из них (LCHa и LCHb) были ассоциированы с мутациями LIS1, DCX и RELN генов. Генные мутации для прочих четырёх подтипов (LCHc, d, e and f) остаются не идентифицированными. Фенотипические проявления заболеваний включают микроцефалию, мальформации коры больших полушарий различные по тяжести проявлений (от агирии до уплощения паттерна извитости при нейровизуализации). Аномалии мозжечка проявляются как в виде срединной гипоплазии его структур, так и в виде выраженной гипоплазии в сочетании с дисплазией.
Таким образом, LCH в рамках спектра генетических мутаций DCX и LIS1 ассоциирована не только с мутацией гена RELN вызывающего тяжёлую церебеллярную и гиппокампальную дисплазию, но может быть вызвана другими, не установленными в настоящее время генетическими мутациями. Детальный анализ клинических, нейровизуализационных признаков у пациентов с различными формами синдромальной лиссэнцефалии проведён Richard Leventer (2008) (табл.). Гипоплазия червя мозжечка определяется у 20% пациентов с LIS (изолированной лиссэнцефалической последовательностью), гипоплазия/дисплазия червя и полушарий мозжечка характерна для всех форм LCH (лиссэнцефалии с церебеллярной гипоплазией) и периодически наблюдается при MLIS (микролиссэнцефалии).
Таблица 2.
Clinical, imaging and genetic features of the main lissencephaly syndromes
Syndrome
Main clinical features
Imaging features
Inheritance
Gene defect
MDS
Facial dysmorphism
Profound global developmental delay
Hypotonia
Intractable epilepsy
Feeding problems
Death within first 2 years
Severe LIS (grade I or 2) with a P>A gradient
Moderately dilated lateral ventricles
AD (sporadic or related to parental balanced translocation involving 17p13.3)
Deletions of 17p13.3 including LISI and 14-3-3є
ILS (LISI)
Global developmental delay (usually severe)
Intractable epilepsy
Evolving spasticity
Mild to severe LIS (grades 2-1) with a P>A gradient
Mildly dilated lateral ventricles
AD (sporadic or related to parental balanced translocation involving 17pl3.3)
X-linked - sporadic or familial*
Deletions or intragenic mutations of LISI
ILS(OCX)
Global developmental delay (usually were) Intractable epilepsy Evolving spasticity
Moderate to severe LIS (grades 1-3) with an A>P gradient Mildly dilated lateral ventricles Mild cerebellar vermis hypoplasia (?20%)
Intragenic mutations of DCX
LCH
Variable clinical features, however most have severe disability with epilepsy, spasticity and often neonatal death RELN LCH - severe global developmental delay, hypotonia, epilepsy
Cerebellar vermis and hemisphere hypoplasia
Variable lissencephaly grade and gradient
Occasional brainstem hypoplasia RELN mutations: A>P gradient and abnormal hippocampi
AD, AR or X-linked – sporadic and familial forms
Most causes unknown LISI DCX or RELN intragenic mutations reported
XLAG
Abnormal or ambiguous genitalia (male)
Temperature instability
Neonatal seizures
Death in neonatal period or infancy
Complete agenesis of the corpus callosum Occasional interhemispheric cyst Mild LIS with a P>A gradient
X-linked - sporadic or familial
Intragenic mutation of ARX
MLIS
Congenital microcephaly <3SD below mean
Variable clinical features including other congenital malformations Neonatal death common
Variable grade and gradient of LIS Occasional periventricular gray matter heterotopia, corpus callosum, brainstem or cerebellar abnormalities
AR sporadic
Unknown
* – мать-носительница может иметь субкортикальную ленточную гетеротопию; † – мать-носительница может иметь агенезию мозолистого тела;
MDS – синдром Miller-Dieker, ILS – изолированная лиссэнцефалическая последовательность, LCH – лиссэнцефалия с церебеллярной гипоплазией, XLAG – Х-сцепленная лиссэнцефалия с аномалией гениталий, MLIS – микролиссэнцефалия, LIS – лиссэнцефалия, P>A - более выраженная степень нарушений в задних отделах больших полушарий, A>P- более выраженная степень нарушений в передних отделах больших полушарий.
Рис. 15. Т1 и T2-weighted изображения мальформаций головного мозга по типу лиссэнцефалии с гипоплазией/дисплазией мозжечка обусловленные различными генетическими мутациями (из монографии «Malformations of theNervous System Handbook of Clinical Neurology Series» Harvey B. Sarnat, Paolo Curatolo/ Chapter13. 2008. Lissencephalytype I. Richard Leventer).
Генерализованная дисплазия мозжечка ассоциированная с диффузной полимикрогирией. Полимикрогирия характеризуется нарушением нормального паттерна складчатости коры с формированием маленьких извилин и нарушением цитоархитектоники. Тяжесть клинических проявлений вариабельная и варьирует от фокальной эпилепсии с сохранным интеллектом до умственной отсталости при билатеральной генерализованной полимикрогирии. Причиной заболевания могут являться внутриутробные инфекции, внутриутробная ишемия плода и ряд генетических синдромов, в том числе врождённые нарушения метаболизма (некетотическая гиперглицинемия, глютаровая ацидемия, сидром Целлвегера). Хромосомные нарушения при полимикрогирии включают микроделецию 22q11 хромосомы (Sztriha etal.,2004), 1p36 (Jansen and Andermann, 2005) и прочие мутации (Leventer et al., 2001). Полимикрогирия часто ассоциируется с другими мальформациями ЦНС, в том числе, диффузной дисплазией полушарий мозжечка (Ben-Omran and Teebi, 2005). Недавно описана мутация в KIAA1279 у пациента с аутосомно-рецессивным заболеванием - Goldberg–Schprintzen syndrome, характеризующимся болезнью Гиршпрунга и билатеральной генерализованной полимикрогирией (Brooks et al., 2005). Идентифицирован ген GPR56, мутация которого ведёт к возникновению аутосомно-рецессивной бифронтальной париетальной полимикрогирии (Piao et al., 2002, 2004). Некоторые формы билатеральной фронтальной полимикрогирии и билатеральной генерализованной полимикрогирии являются Х-сцепленными (Villard et al., 2002).
Рис.16. Кортикальная дисплазия мозжечка у 3-х месячного ребёнка с врождённой цитомегаловирусной инфекцией ассоциированная с полимикрогирией в левом полушарии большого мозга (Patel S, Barkovich J, 2003).
Фокальная дисплазия. Изолированная дисплазия червя
Joubert Syndrome описан впервые Joubert et al в 1969 году у четырех сибсов. В 1997 году Maria обратил внимание на симптом «коренного зуба» (molar tooth sign), выявляемый при МРТ головного мозга, однако, уже тогда было ясно, что данный симптом не является строго специфичным (Satran et al., 1999).
Joubert Syndrome включает 6 вариантов аномалии, различных в зависимости от варианта генных мутаций. В настоящее время установлены мутации в следующих генах: AHI1, CORS2, JBTS1, TMEM67, NPHP1, CEP290. В 10% случаев синдрома Joubert цитогенетическое исследование выявляет мутации в генах NPHP1, CEP290, AHI1 и TMEM67(MKS3).Морфологическими признаками синдрома являются отчётливая аномалия мозжечка и ствола мозга. Клинические признаки включают мышечную гипотонию, расстройство психомоторных функций, эпизоды апноэ и тахипноэ, у большинства детей определяется туловищная атаксия. Общим признаком является нарушение развития статикомоторных функций (Saraiva & Baraitser 1992; Steinlin et al 1997; Maria, Bolthauser et al 1999; Bennett et al 2003). Степень когнитивных расстройств вариабельна, от глубокой умственной недостаточности до нормы. Дыхательные расстройства проходят с возрастом (Maria, Quisling et al 1999). Фенотип при синдроме Joubert вариабелен. К часто встречающимся признакам этого синдрома относятся ретинальная дистрофия, колобома радужки, аномалии почек, фиброз печени, полидактилия, оральные гамартомы и эндокраниальные аномалии. Рядом авторов описаны судорожные приступы у пациентов с этим синдромом (Saraiva & Baraitser 1992). Частота встречаемости синдрома в популяции оценивается от 1:258 000 (Flannery & Hudson 1994) до 1:100 000 по последним данным (D Flannery and MParisi).
Диагноз синдрома Joubert базируется на наличии характерных клинических признаков и симптома «коренного зуба» на МРТ головного мозга. Корректное проведение исследования требует выполнения срезов через средний мозг и мост. Характерный вид «коренного зуба» вызван следующими структурными аномалиями (Maria, Quisling et al 1999; Shen WCet al, 1990):
При проведении аксиальных срезов:
При проведении коронарных срезов:
При проведении сагиттальных срезов:
При проведении парасагиттальных срезов:
Рис. 17. Результат МРТ ребёнка с Joubert Syndrome.
Сопутствующие аномалии ЦНС могут быть представлены аномалией заднечерепной ямки по типу вариантаDandy-Walker, которая определяется у 10% пациентов с Joubert-синдромом (Maria et al 2001), затылочным энцефало- или менингоцеле (Genel et al 2004), гидроцефалией, требующей оперативного вмешательства с целью шунтирования без признаков классического синдрома Dandy-Walker (Genel et al 2004), агенезией мозолистого тела (Valente et al 2005), нейроэпителиальными кистами (Marsh et al 2004), полимикрогирией (Gleeson et al 2004, Dixon-Salazar et al 2004), гетеротопией (Saraiva & Baraitser 1992).
Внешний вид пациентов с синдромом Joubert характеризуется удлиненным лицом с высокорасположенными изогнутыми бровями, птозом, седловидным носом, треугольным острым ртом, прогнатизмом. Следует отметить, что эти черты встречаются при ряде других синдромов, и, кроме того, не чётко выражены в младенчестве, однако, многие авторы описывают «Joubert facies» (Maria, Bolthauser et al 1999, Braddock et al 2003, 2006).
Следует отметить, что симптом «коренного зуба» на МРТ изображении встречается при описании ряда синдромов, имеющих общие признаки с синдромом Joubert. Это свидетельствует об отсутствии на данный момент чёткого представления о диагностических критериях патологии и требует дальнейшего обсуждения для формирования общих взглядов и подходов к диагностике, а также продолжения молекулярно-генетических исследований.
Постановка диагноза синдрома Joubert требует проведения высокопольной МРТ с шириной шага в три миллиметра, выполнения стандартных аксиальных, коронарных и сагиттальных проекций, а также аксиальной проекции через средний мозг, мост и заднечерепную ямку.
Таблица 3. Молекулярная генетика синдрома Joubert
Locus Name
Gene Symbol
Chromosomal Locus
Protein Name
CORS2 (JBTS2)
Unknown
11p12-q13.3
Unknown
JBTS1
Unknown
9q34.3
Unknown
JBTS3
AHI1
6q23.3
Jouberin
JBTS4
NPHP1
2q13
Nephrocystin-1
JBTS5
CEP290
12q21.32
Centrosomal protein Cep290
JBTS6
TMEM67
8q21.1-q22.1
Meckelin
Таким образом, Joubert-синдром представляет собой врождённое аутосомно-рецессивное заболевание. Риск развития патологии для сибсов составляет 25%, в 50% сибсы могут являться носителями патологического гена, и в 25% могут не иметь патологического гена.
Rhombencephalosynapsis (RES) – редкая врождённая аномалия мозжечка характеризующаяся гипоплазией или агенезией червя мозжечка, слиянием по средней линии его гемисфер с поперечным расположением борозд, слиянием верхних и/или средних ножек и зубчатых ядер, отсутствием ядер шатра, их афферентных и эфферентных связей, глубокие ядра мозжечка, как правило, сформированы. Аутопсийное исследование выявляет уменьшение поперечного размера мозжечка. Вид конвекситальной поверхности мозжечка, разделение полушарий на доли не нарушены, на передней и задней поверхности присутствуют все основные борозды, нормально ориентированные извилины полушарий спаяны по средней линии. Отсутствует incisura cerebelli posterior, в норме располагающаяся позади червя мозжечка, также отсутствует передний мозговой парус. Верхние ножки мозжечка спаяны и расположены над Сильвиевым водопроводом. Ядра блоковидных нервов могут быть смещены латерально, в одном случае описано отсутствие ядер тройничного нерва (Gross H, 1959), часто выявляется нарушение структуры оливарного комплекса: могут отсутствовать дорзальные и медиальные добавочные ядра олив, нижние ядра олив при этом остаются не измененными (Gross H, 1959; Schachenmayr W, Friede RL, 1982). Разветвление водопровода описано Kepes JJ et al (1969), сужение водопровода с глиозом окружающих тканей выявлено Isaac M, Best P (1987). Гистологическое исследование может выявить гетеротопию клеток Пуркинье, описанную в трёх источниках. Аномалии ориентации борозд коры мозжечка, описаны, но не типичны и ведут к диагностическим трудностям (Soto et al., 2004). Takanashi et al. (1999) описал случай RES с вертикальной ориентацией борозд полушарий мозжечка и мультифокальными кистоподобными из-менениями коры мозжечка.
Первое описание rhombencephalosynapsis было опубликовано Obersteiner в 1914 году и заключалось в описании аномалии мозга обнаруженной при вскрытии трупа 28 летнего покончившего с собой мужчины. Термин «rhombosynapsis» был впервые применён De Morsier (1955) как противопоставление другой мальформации представляющей собой «расщелину» в полушариях мозжечка и определяемой термином «rhomboschizis», Gross и Hoff в 1959 году предложили сохранить термин для определения порока представленного слиянием гемисфер мозжечка. До 1995 году было описано 18 случаев этой редкой патологии, выявленных при патологоанатомическом вскрытии. Публикации о прижизненной ди-агностике rhombencephalosynapsis с помощью методов нейровизуализации появились с 1991 года (Savolaine et al., 1991; Truwit et al., 1991). Всего в литературе описано 58 случаев данной аномалии. В настоящее время возможна пренатальная диагностика RES с помощью методов нейровизуализации (МРТ) (Napolitano M. еt al, 2004). Sener (2000), проведя 3000 МРТ-исследований структур головного мозга у детей, оценил частоту RES как 0,13%.
Рис. 18. Rhombencephalosynapsis у 6-и летнего мальчика (Patel S, Barkovich AJ,2003).
В настоящее время, ведущее место как метод диагностики RES имеет магнитно-резонансное исследование головного мозга, позволяющее оценить структуру червя мозжечка, его разделение на дольки, оценить границу между серым и белым веществом, расположение ядер мозжечка. МР-признаки патологии при проведении аксиального среза включают ромбовидную форму IV желудочка, вместо серповидной формы в норме, что связанно с гипоплазией или агенезией червя. Позади IV желудочка могут определяться спаянные зубчатые ядра и верхние ножки мозжечка, спаянные гемисферы мозжечка. Коронарные срезы позволяют выявить продолжение через среднюю линию борозд полушарий при отсутствии между ними червя мозжечка.
Большинство случаев RES ассоциированы с супра- и субтенториальными мальформациями. Аномалия может ассоциироваться с вентрикуломегалией, отсутствием прозрачной перегородки, дисгенезией мозолистого тела (Truwit et al., 1991; Simmons et al., 1993; Demaerel et al.,1995; Montull et al., 2000), слиянием структур свода и ядер таламуса (Simmons et al, 1993; Kepes et al., 1969). Вентрикуломегалия описана в 39 из имеющихся 58 случаев. Возможен стеноз водопровода с развитием окклюзи-онной гидроцефалии. Для лечения 14 пациентов потребовалось проведение вентрикулоперитонеального шунтирования (Schachenmayr et Friede, 1982; Truwit et al., 1991; Simmons et al., 1993; Von Boltenster n et al., 1995; Utsonomi ya et al., 1998; Danon et al., 2000; Guyot et al., 2000; Oei et al., 2001; Toelle et al., 2002; Yachnis, 2002). Преобладают аномалии вентральной индукции, формирующиеся параллельно развитию мозжечка в нейроонтогенезе. Голопрозэнцефалия ассоциированная с RES наблюдалась в ряде случаев (Garfinkle, 1996; Sergi et al., 1997; Siebert et al., 2005). В литературе описана гипоплазия височных долей (Montull et al., 2000; Truwit et al., 2001), септооптическая дисплазия с гипоплазией оптических нервов, хиазмы зрительных нервов, и зрительных трактов (De Morsier, 1956; Michaud et al., 1982). Аномалии срединного мозга могут включать, например, спаянные бугорки, агенезию задней доли гипофиза. Задняя черепная ямка обычно меньше, чем в норме. В двух случаях выявлена ассоциация мальформации с энцефалоцеле (Schachenmayr et Friede, 1982; Toelle et al., 2002). Sener RN, Dzelzite S (2003) приводит пример RES ас-социированного с менингомиелоцеле цервикоторокальной локализацией и аномалией Chiari II.
С пороком могут ассоциироваться аномалии мышц, скелета, сердечно-сосудистой, мочевыделительной, дыхательной системы. Описанные в литературе проявления дизморфизма носят неспецифический характер и могут быть представлены такими признаками как низко расположенные ушные раковины, гипертелоризм, высокое нёбо (Truwit C et all, 1991; Toell SP et all, 2002; Romamendo M et all, 1997).
В настоящее время не установлена чёткая корреляция между клиническими проявлениями патологии и МРТ признаками аномалии, нередко порок обнаруживался случайно у взрослых (Savolaine E et all, 1991; Bell BD et all, 2005). Прогноз развития психоневрологических функций у пациентов вариабелен и зависит от тяжести сопутствующих супратенториальных аномалий. Клинические проявления изолированного RES в большинстве случаев представлены умеренной туловищной атаксией, в сочетании с нистагмом и/или сходящимся косоглазием, описаны пирамидные и экстрапирамидные расстройства, развитие когнитивных функций у шести пациентов соответствовало возрасту, у одной женщину опи-сана психиатрическая патология при нормальном интеллекте (Demaerel et al.,1995; Truwit C et all, 1991; Toell SP et all, 2002; Guyot LL et al, 2000; Mendonca JL et al., 2004; Bell et al., 2005).
Большинство случаев мальформации являются спорадическими. У трёх пациентов с RES подтверждены хромосомные нарушения (Truwit et al., 1991; Castillo M., 1996; Lespinasse et al., 2004), пациенты имели умственную отсталость, обусловленную генетическим дефектом.
RES является характерным признаком церебелло-тригемино-дермальной дисплазии - синдрома Gomez–Lopez-Hernandez (Romanengo et al., 1997). Прочие признаки синдрома включают краниостеноз с формированием брахицефалии, анестезию в зоне иннервации тройничных нервов, алопецию с височной и/или теменной локализации, умственную отсталость с нарушением речи, поведения и атаксию. МРТ исследование головного мозга выявляет структурные изменения типичные для RES, вентрикуломегалию.
Описано два случая рождения детей с RES от матерей, страдающих сахарным диабетом (Garfinkle, 1996; Sergi et al., 1997). В двух случаях порок сформировался у женщин получавших антиконвульсанты: этосуксимид и комбинацию клоназепама с вальпроевой кислотой (Demaerel et al., 1995; Toelle et al., 2002).
Изолированная дисплазия гемисфер
Lhermitte-Duclos-Cowden disease. Данное состояние (Lhermitte-Duclos disease) трудно для прижизненной диагностики, оно может дебютировать у пациентов грудного возраста или в раннем детстве в виде увеличения размеров головы и признаков внутричерепной гипертензии. Посмертно при вскрытии обращает внимание увеличение размеров извилин за счёт сосредоточения узлов патологических клеток. Гистологически определяются скопления гигантских изменённых нейронов в гранулярном слое, и множество аномально миелинизированных волокон во внешнем молекулярном слое. Предполагается идентичность этого синдрома с Cowden's disease (Eng et al., 1994), характерными признаками которого является развитие во второй декаде жизни папулезных ново-образований на коже лица, сосредоточенных вокруг анатомических отверстий и ассоциированных с изменением структуры волос. Маленькие гиперкератозные и бородавчатые бляшки расположены также на тыльной стороне кистей и стоп. Часто встречаются папулезные образования на слизистой оболочке рта, характерен складчатый «мошоночный» язык. Большинство пациентов имеют большую окружность головы и широкий лоб. В молочных железах у женщин формируются фиброзно-кистозные образования склонные к малигнизации, для мужчин характерны аденоматозные полипы желудочно-кишечного тракта. Hanssen and Fryns (1995) в трети случаев при Cowden syndrome у детей описывают прогрессирующую макроцефалию с умеренной задержкой развития психомоторных функций. Padberg et al., (1991) предположил, что данное состояние и Lhermitte-Duclos disease являются проявлением одного и того же заболевания. В обоих случаях описаны гамартоматозные поврежде-ния мозжечка, характеризующиеся наличием ганглиев гипертрофированных нейронов в гранулярном и молекулярном слоях и чрезмерного утолщения миелиновой оболочки аксонов. Eng et al., (1994) описал семью четыре члена которой имели признаки Cowden syndrome и Lhermitte-Duclos disease. Nelen et al., (1996) картировал ген на 10q22-23.
Рис. 19. Фокальная церебеллярная дисплазия у ребёнка грудного возраста (Patel S, Barkovich AJ., 2003).
Коронарное изображение позволяет увидеть регион фокальной дисплазии (стрелки) в нижнемедиальном отделе левой гемисферы мозжечка.
Прочие фокальные дисплазии. Описан случай фокальной дисплазии полушария мозжечка у 12-летнего мальчика, обратившегося для обследования в связи травмой головы. Фокальная дисплазия гемисферы и червя мозжечка была выявлена у 10-месячного младенца с нарушением двигательного развития (Patel S, Barkovich AJ, 2003). В обоих случаях повреждение визуализировалось в виде нарушения архитектоники в области медиальной поверхности гемисферы в одном случае и медиальной поверхности гемисферы и смежной поверхности червя в другом случае. Регион дисплазии имел ту же интенсивность МРТ сигнала, что и кора мозжечка, но сигнал от диспластичного белого вещества мозжечка несколько отличался и был гиперинтенсивным в режиме T2-weighted изображения по сравнению с неизменённым белым веществом полушарий мозжечка. Объём мозжечка был несколько уменьшен в районе дисплазии.
Цель приведённой классификации формирование общих представлений у неврологов, молекулярных биологов о закономерностях развития мозжечка и закономерностях нарушения его развития. Классификация удобна для описания пороков развития при проведении нейровизуализации, однако, с точки зрения клинициста нельзя не отметить, ряд недостатков:
Таким образом, проблема классификации нарушений развития ЦНС сохраняет своё значение, порок развития отдельно взятой структуры должен рассматриваться элемент нарушения нейроонтогенеза и в рамках нарушения нейроонтогенеза. Исследование физиологической роли мозжечка в функционировании нервной системы не может основываться только на анализе и классификации морфологических и гистологических типов расстройств, но, при современном уровне знаний, должно учитывать варианты нарушения нейротрансмиссии.
Агенезия мозолистого тела.
Агенезия мозолистого тела
Imaging in Dandy-Walker Malformation
http://emedicine.medscape.com/article/408059-overview#showall